АМИЛОИДОЗ
Расстановка ударений: АМИЛОИДО`З
Содержание
Амилоид и его свойства Этиология и патогенез Патологическая анатомия Клиническая картина Прогноз Диагноз Лечение Амилоидоз кожи Амилоидоз у детей
Амилоидоз (amyloidosis; греч. amylon — крахмал, eidos — вид + -ōsis), амилоидная дистрофия — нарушение белкового обмена, выражающееся в отложении и накоплении в тканях белковых веществ с характерными физ.-хим. свойствами. Этиологически и патогенетически объединяет разные процессы, ведущие к образованию в тканях сложного глюкопротеида — амилоида.
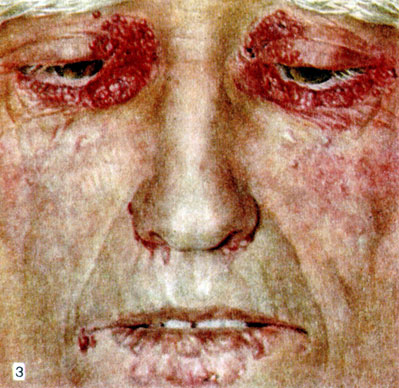
Первичный Системный амилоидоз кожи
Изучению А. способствовали описания Рокитанским (С. Rokitansky, 1844) «сальной болезни» и Вирховом (R. Virchow, 1853) амилоида, а также создание экспериментальной модели амилоидоза Н. П. Кравковым и Кучинским (М. N. Kuczynski). Наиболее распространена казеиновая модель А., получаемая путем введения мышам или кроликам подкожно 5—10% взвеси или раствора казеината натрия. Однако экспериментальный А. можно вызвать также с помощью культуры золотистого стафилококка, синегнойной и дифтерийной палочек, гонококка, холерного вибриона, скипидара, протеолитических ферментов, коллоидных растворов серы, селена и др.
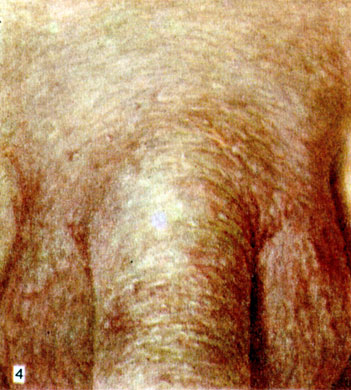
Первичный Системный амилоидоз кожи
Распространение А. в различных странах неодинаково. Частота его в Испании — 1,9% вскрытий, в Португалии — 1,4%, Израиле — 0,55%, в Японии — только 0,1%, что нек-рые авторы склонны объяснять особенностями питания населения.
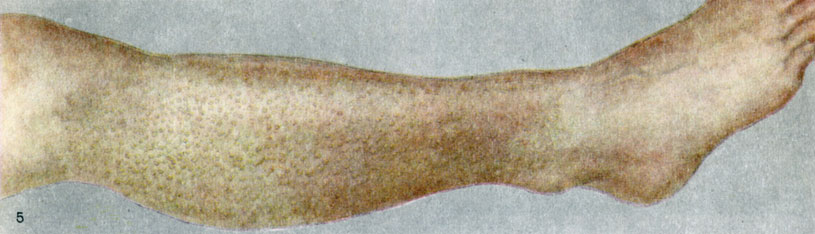
Амилоидный лихен
Наиболее частая локализация А. — почки, где он обнаруживается, по данным А. А.Демина (1970), в 1,4% случаев, по данным Г. П. Шульцева (1970), охватывающим секционные наблюдения 60—70-х годов, — в 1,9% случаев.
Амилоид и его свойства
Амилоид имеет сложное строение. Основным компонентом его являются белки, среди к-рых обнаружены как фибриллярные (тканевые) белки типа коллагена, так и плазменные белки — α- и γ-глобулины, фибриноген. Полисахариды амилоида представлены хондроитинсерной и гиалуроновой кислотами, гепарином, нейраминовой к-той, причем преобладают хондроитинсульфаты. Амилоид обладает антигенными свойствами; устойчив к действию многих ферментов, кислот, щелочей благодаря прочности связей между белковыми и полисахаридным компонентами.
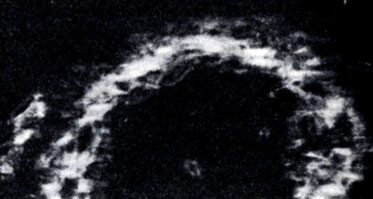
Рис. 1. Амилоидоз селезенки. Положительная анизотропия амилоидных отложений
Хим. состав амилоида неодинаков при различных формах и типах А., чем объясняется различное его отношение к красителям (конго красный, метил- или генцианвиолет, йод и йод-грюн) и разная интенсивность характерной метахроматической реакции; в ряде случаев эта реакция отсутствует (ахроматический амилоид, или ахроамилоид, параамилоид). Наиболее специфичной для амилоида является его люминесценция с тиофлавином S или Т.
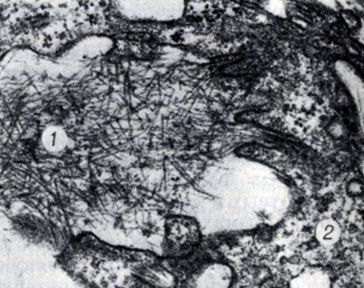
Рис. 2. Фибриллы амилоида (1) по периферии ретикулярных клеток (2) в связи с инвагинациями цитоплазматической мембраны (культура тканей; × 53000)
Амилоид имеет фибриллярную паракристаллическую структуру. В связи с этим он обладает дихроизмом и анизотропией (см.); последняя наиболее отчетливо выражена при окраске конго красным (рис. 1). Спектр положительного двойного лучепреломления амилоида лежит в пределах 540—560 нм. Эти поляризационно-оптические свойства амилоида позволяют отличать его от коллагена, ретикулина, эластина.
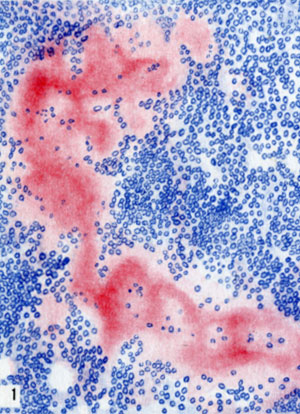
Амилоидоз селезенки. Отложение амилоида по периферии фолликула (окраска красным конго)
Фибриллы амилоида, выявляемые при электронномикроскопическом исследовании, имеют диаметр 7,5 нм и длину до 800 нм; они лишены поперечной исчерченности. Каждая фибрилла состоит из двух субфибрилл (филаментов) по 2,5 нм в диаметре, к-рые расположены параллельно на расстоянии 2,5 нм друг от друга. Установлено внутриклеточное образование фибрилл амилоида, а также причастность к фибриллогенезу мезенхимальных клеток — ретикулярной клетки, фибробласта (рис. 2), что позволило считать амилоид фибриллярным аномальным белком.
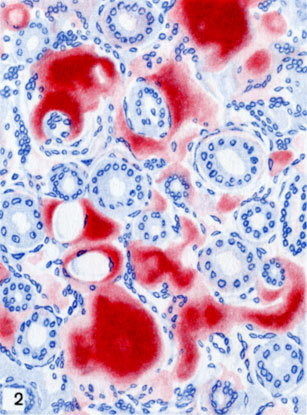
Отложение амилоида в мозговомвеществе почки (окраска красным конго)
В отличие от коллагена, структурные белки фибрилл амилоида богаты триптофаном и не содержат гидроксипролина, связаны с небольшим количеством нейтральных Сахаров и сиаловой к-ты.
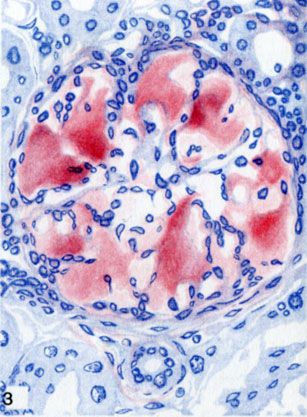
Отложение амилоида в капиллярныхпетлях клубочка почки (окраска красным конго)
Структурно-химические свойства фибрилл определяют специфическое окрашивание амилоида конго красным, молекулы к-рого прочно удерживаются между филаментами фибриллы водородными связями сульфогрупп красителя с основными группами белка.
Помимо фибрилл, в амилоиде выделены специфические палочковидные структуры («периодические палочки», или Р-компонент) диаметром 10 нм и длиной до 400 нм. Они состоят из отдельных пентагональных образований диаметром 9—10 нм, расположенных друг от друга на расстоянии 4 нм. Каждое такое образование представлено пятью триангулярными компонентами до 2,5 нм в диаметре. Палочковидные структуры относятся к глюкопротеидам сывороточного происхождения, в них по сравнению с фибриллами амилоида значительно выше содержание нейтральных Сахаров и сиаловой к-ты; они определяют антигенные свойства амилоида.
Этиология и патогенез
Этиология и патогенез А. не вполне ясны. Обсуждаются три основные теории патогенеза А.
I. Теория локального клеточного генеза Тайлума (G. Teilum, 1954) объясняет амилоидогенез лишь на уровне клетки. При этом речь идет о синтезе клеткой ретикулоэндотелия не амилоида — сложного гетерогенного вещества, состоящего из фибриллярных и нефибриллярных структур, а лишь фибриллярных его предшественников. Амилоид же образуется вне клетки в тесной связи с волокнистыми структурами соединительной ткани. Согласно этой теории в амилоидогенезе можно выделить две фазы: пред амилоидную и собственно амилоидную. До появления амилоида в ретикулоэндотелиальной системе наблюдается пролиферация и плазмоцитарная трансформация с образованием пиронинофильных клеток, богатых РНК. В собственно амилоидную фазу происходит подавление пролиферации клеток ретикулоэндотелия, истощение пиронинофильных клеток, появление клеток, богатых полисахаридами (PAS-клетки), которые «строят» амилоидную субстанцию. Теория «локального клеточного генеза» объясняет ряд фактов, известных из клинико-экспериментальных наблюдений. С позиций этой теории можно объяснить, в частности, избирательность поражения элементов ретикулоэндотелиальной системы при вторичном А., прежде всего областей, наиболее активных в функциональном отношении: маргинальных зон фолликулов селезенки, синусоидов печени. Существенным аргументом в пользу теории локального клеточного генеза являются работы, в к-рых убедительно показано образование амилоида в культуре ткани. Однако доказательства интрацеллюлярного образования фибрилл амилоида клеткой ретикулоэндотелия (PAS-клеткой) не решают вопроса в пользу признания теории «локального клеточного генеза» универсальной.
II. Согласно иммунологической теории Лешке — Леттерера (Н. Loeschke, Е. Letterer, 1927) образование амилоида рассматривается как результат реакции антиген — антитело, где антиген — продукт распада тканей или чужеродный белок, а амилоид — белковый преципитат, откладывающийся прежде всего в местах образования антител. Амилоид возникает при условии плохой выработки антител и избытке антигена. В пользу иммунологической теории свидетельствуют гипергаммаглобулинемия в пред амилоидную стадию, падение иммуноглобулинов в период образования амилоида, характер морфологических изменений в органах и т. д. Однако эта теория не объясняет случаев развития А. у лиц с гипо- и агаммаглобулинемией. Участие иммунологических механизмов в развитии А. требует дальнейшего изучения.
III. Теория диспротеиноза, или органопротеидоза Кагли (V. Cagli, 1961), рассматривает амилоид как продукт извращенного белкового обмена. С позиций этой теории основное звено в патогенезе А. — диспротеинемия с накоплением в плазме грубодисперсных белковых фракций и аномальных белков. Гиперфибриногенемия может быть также условием образования амилоида.
Патологическая анатомия
Амилоидные отложения имеют типичную локализацию в стенках кровеносных и лимф, капилляров и сосудов, в интиме или в адвентиции; в строме органов по ходу ретикулярных или коллагеновых волокон; в собственной оболочке железистых структур.
В зависимости от отношения амилоида к различным клеткам (ретикулярная клетка, фибробласт) или различным волокнам соединительной ткани (ретикулярные, коллагеновые), среди к-рых амилоид выпадает, различают периретикулярный и периколлагеновый А.
Для периретикулярного А., при к-ром амилоид выпадает по ходу содержащих ретикулин мембран сосудов и желез, а также ретикулярной стромы паренхиматозных органов, характерно преимущественное поражение селезенки, печени, почек (цветн. табл., ст. 73, рис. 1—3), надпочечников, кишечника, интимы сосудов мелкого и среднего калибра (так наз. паренхиматозный, типичный А.). Для периколлагенового А., при к-ром амилоид выпадает по ходу коллагеновых волокон, характерно преимущественное поражение адвентиции сосудов среднего и крупного калибра, миокарда, поперечнополосатой и гладкой мускулатуры, нервов, кожи (так наз. мезенхимальный, системный А.).
Незначительные отложения амилоида, выявляющиеся лишь при микроскопическом исследовании, обычно не ведут к функциональным нарушениям (клинически латентная стадия А.) и не изменяют внешний вид органов.
Прогрессирующий А., как правило, ведет к функциональной недостаточности органа, что связано с атрофией его паренхиматозных элементов и склерозом. Орган увеличивается в объеме, становится плотным т ломким, имеет своеобразный восковидный или сальный вид на разрезе («сальная селезенка», «восковидная печень»). В финале развивается амилоидное сморщивание органа, напр. амилоидное сморщивание почек (рис. 3). Т. о., разнообразие причин возникновения А. и механизмов образования амилоида делает малооправданными поиски единой теории амилоидогенеза. Нет, очевидно, единого амилоидоза как клинико-морфологического понятия, есть амилоидозы. Конкурирующие теории А. объясняют лишь отдельные звенья его патогенеза (гуморальные, тканевые, клеточные).
Однако имеется ряд признаков, общих для всех форм А. К ним относятся: диспротеинемия, являющаяся выражением нарушенного белкового обмена и процесса обновления белков организма; трансформация клеток ретикулоэндотелиальной системы с возникновением фибриллярной структуры амилоида; типичные субмикроскопические изменения, предшествующие появлению амилоида; единая субмикроскопическая структура амилоида.
Общепринятой классификации А. не существует. Любарш (О. Lubarsch, 1929) и Рейманн (Н. A. Reimann, 1935) выделили первичный (идиопатический) и вторичный амилоидоз. Бриггс (G. W. Briggs, 1961) различает следующие виды А.: 1. Первичный: а) генерализованный; б) семейный; в) респираторный — опухолевидный (узловатый) и диффузный. 2. Вторичный. 3. Старческий кардиальный. 4. А. при миеломной болезни. 5. Опухолевидный локальный А. (исключая респираторный).
По Хеллеру (Н. Heller, 1966), существуют три группы А., в каждой из к-рых выделяется ряд форм:
I. Генетический (наследственный) А.: 1) семейная средиземноморская лихорадка (периодическая болезнь); 2) семейный А. с лихорадкой, крапивницей и глухотой (форма Маккла — Уэллса); 3) нейропатический с преимущественным поражением нижних или верхних конечностей; 4) кардиопатический.
II. Приобретенный А.: 1) как осложнение хроя. инфекций (бронхоэктатическая болезнь, туберкулез, остеомиелит), коллагеновых болезней (ревматоидный артрит и др.), злокачественных опухолей; 2) как проявление множественной миеломы.
III. Идиопатический А.: 1) классический первичный; 2) нефропатический; 3) нейропатический; 4) кардиопатический; 5) локализованный.
При различных формах в каждой группе тип отложения амилоида (периретикулярный или периколлагеновый) может быть разным.
Классификация Хеллера по сравнению с классификацией Бриггса более прогрессивна. Она более строго очерчивает группу идиопатического А., включая в нее, помимо генерализованной формы первичного А., различные клинические его варианты с преимущественным поражением почек, нервной системы, сердца. Оправдано выделение в самостоятельную группу генетического А., происхождение к-рого связывают с наследственной ферментопатией, определяющей дефект синтеза фибриллярных белков организма. Среди форм наследственного А. наиболее распространенным является А. при периодической болезни (семейной средиземноморской лихорадке), особенно часто описываемый у армян, евреев, арабов и наследуемый по аутосомно-рецессивному типу. При этом А. может быть единственным проявлением этого генетически обусловленного страдания (II фенотип) или развиваться вслед за периодом лихорадки и приступов болей (I фенотип), приводя и в том, и в другом случае к почечной недостаточности. Выделяют также так паз. португальский амилоидоз, протекающий с преимущественной периферической нейропатией и передающийся по доминантному типу, и А. с преимущественным поражением сердца (кардиопатический), заканчивающийся смертью от сердечной недостаточности.
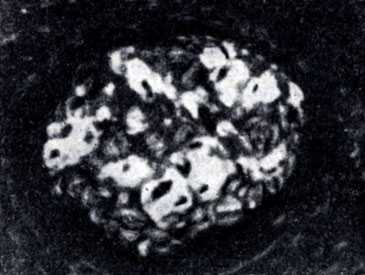
Рис. 3. Амилоидно-сморщенная почка. Амилоид (светлые участки) в капиллярных петлях клубочка (люминесценция с тиофлавином Т)
Гафни (J. Gafni, 1964) сводит все разнообразие наследственного А. к трем клинико-морфологическим типам (формам): 1) нефропатический, проявляющийся протеинурией, нефротическим синдромом и уремией в финале заболевания; 2) нейропатический, характеризующийся прогрессирующим полиневритом с мышечной атрофией, импотенцией, кишечными расстройствами и кахексией; 3) кардиопатический, к-рому свойственна нарастающая сердечная недостаточность.
Частой причиной приобретенного (вторичного) А. являются хрон. специфические инфекции (туберкулез, сифилис), особенно хрон. нагноения (остеомиелит, бронхоэктатическая болезнь), ревматоидный артрит, реже неспецифический язвенный колит, лимфогранулематоз, опухоли («cancer-амилоидоз»).
В группу вторичного А. относят также и периколлагеновые отложения так наз. параамилоида при миеломной болезни и макроглобулинемии Вальденстрема, связанные с извращенным синтезом глобулинов опухолевыми клетками.
Показано, что в развитии вторичного А., осложняющего хрон. инфекции, коллагеновые болезни, хрон. гломерулонефрит, велика роль иммунологических (аутоиммунных) реакций. Механизм образования параамилоида сводится к отложению в ткани парапротеина, к-рый вступает в реакцию белок — белок или белок — полисахарид, что ведет к преципитации нерастворимого параамилоида.
Локализованный (местный) А., к-рый относят к первичному, имеет нек-рые особенности. Выпадая в слизистой оболочке носа, глотки, голосовых связок, трахеи и бронхов, в стенке мочевого пузыря и мочеточников, в клетчатке век и языке, а также коже, амилоид образует опухолевидные структуры (опухолевидный А.).
Классификацию Хеллера следует дополнить старческим А. (группа приобретенного А.). У старых людей наиболее часто встречается А. мозга, сердца и островков Лангерганса, что, по Швартцу (P. Schwartz, 1970), составляет характерную патологоанатомическую триаду, обусловливающую старческую психическую и физическую деградацию. Амилоидные отложения в других органах и тканях старых людей встречаются значительно реже. Швартц рассматривает старческий амилоидоз как болезнь возраста и считает, что у старых людей имеется несомненная связь А., атеросклероза и диабета, к-рые объединяют единые метаболические нарушения.
Клиническая картина
Клиническая картина А. многообразна и зависит от локализации и интенсивности амилоидных отложений в органах, длительности заболевания, наличия осложнений. Особенно демонстративной она становится при поражении почек, сердца, нервной системы, кишечника.
Амилоидоз почек. Почки поражаются часто как при вторичном А., так и при первичном. Постепенное распространение амилоидных отложений с вовлечением сосудистой стенки приводит к нарастающей протеинурии с возникновением далее нефротического синдрома, снижением почечного кровотока, клубочковой фильтрации, развитием нефрогенной гипертонии и азотемии. При этом могут наблюдаться, а иногда и преобладать проявления основного заболевания, приведшего к А., — ревматоидного артрита, периодической болезни, лимфогранулематоза и др., что затрудняет распознавание А.
Больные А. почек жалуются на общую слабость, снижение аппетита, отеки, часто впервые возникающие на ногах; в дальнейшем они распространяются по всему телу, затрудняют дыхание, пищеварение, мочеотделение. Бывают боли в области поясницы, особенно резкие и острые , при тромбозе почечных вен. Развиваются артериальная гипертония, почечная недостаточность.
При А. олигурия периода больших отеков может смениться в стадии хрон. почечной недостаточности полиурией, но нередко олигурия вместе с отеками сохраняется и в терминальном периоде болезни. Иногда возникает диарея. При значительном поражении дистальных канальцев может наблюдаться избыточное выделение с мочой натрия и воды, так же как при А. надпочечников (нефрогенный инсипидарный синдром), и канальцевый метаболический ацидоз.
Для А. почек характерна протеинурия (см.), развивающаяся при всех формах заболевания, но наиболее выраженная при вторичном А. При этом за сутки выделяется от 2 до 20 и даже 40 г белка, в основном альбумина и в меньших количествах — глобулинов, гликопротеидов, особенно α1-гликопротеидов, и др. Значительная протеинурия сохраняется при развитии хронической и даже терминальной почечной недостаточности.
Продолжительная потеря белка почками, нередко вместе с меньшим приемом его с пищей, пониженным всасыванием, а иногда и усиленным выведением его через жел.-киш. тракт, так же как и повышенный белковый катаболизм, приводит к гипопротеинемии и отекам.
Как правило, отеки развиваются довольно рано и приобретают распространенный и упорный характер. Особенно постоянно снижение содержания в крови альбуминов и повышение α2- и γ-глобулинов — диспротеинемия, усиливаемая и основным заболеванием (активный туберкулез, обострение ревматоидного артрита).
Имеет место повышение содержания гликопротеидов и липопротеидов соответственно в α1- и β-фракциях с одновременным снижением гликопротеидов в альбуминах, изменяется содержание сывороточных иммуноглобулинов и снижается титр комплемента. Часто резко ускоряется РОЭ и изменяются осадочные пробы. Характерна гиперлипидемия с повышенным содержанием холестерина и значительным преобладанием β-липопротеидов крови. Гиперхолестеринемия сохраняется обычно и у истощенных больных (напр., при кавернозном туберкулезе), так же как нередко и в уремической стадии вместе с резко выраженной протеинурией и отеками.
Сочетание массивной протеинурии, гипопротеинемии, диспротеинемии, гиперхолестеринемии и отеков составляет весьма характерный для А. почек классический нефротический синдром, наблюдаемый в среднем у 60% больных вслед за нередко весьма длительной латентной стадией умеренной протеинурии. Появление развернутого нефротического синдрома может быть спровоцировано интеркуррентной инфекцией, охлаждением, травмой, вакцинацией, лекарственными воздействиями, обострением основного заболевания.
При А. встречается также гиперфибриногенемия, гиперкоагуляция с развитием тромбоза почечных вен и др. Можно обнаружить также лейкоцитоз в крови, анемию, костномозговой плазмоцитоз, синдром нефрогенного диабета.
Сравнительно часто выявляется стойкая микрогематурия, а иногда и макрогематурия, так же как и лейкоцитурия, без сопутствующего пиелонефрита. Обнаруживают и липоидурию с наличием двоякопреломляющих кристаллов в осадке мочи.
По мнению ряда клиницистов, наиболее типичным для А. почек является переход раннего, безотечного периода с незначительной протеинурией в отечную стадию, а затем в кахектический или уремический период. А. почек повторяет в общих чертах эволюцию классической болезни Брайта.
Стадии хрон. почечной недостаточности (см.) свойственны все основные черты нарушения концентрационной и азотовыделительной функции почек — падение удельного веса мочи, снижение клубочковой фильтрации, задержка выведения креатинина и других азотистых шлаков. Сморщенная почка может быть выявлена также с помощью обзорной рентгенограммы, сканирования, методики пневморена.
Артериальная гипертензия при А. развивается чаще параллельно со стойкой азотемией, анемией, т. е. в конечной стадии болезни как проявление диффузного поражения почек, но может быть и в начале нее, что связано с поражением сосудов почек.
Длительное существование артериальной гипертензии приводит к гипертрофии левого сердца с развитием сердечной недостаточности. Вовлекаются сосуды глазного дна. Однако злокачественная гипертония со слепотой и резко измененной ЭКГ при А. редка. При А. возможен также коллапс в результате присоединения инфекционных и тромботических осложнений или прогрессирующая артериальная гипотензия вследствие А. надпочечников.
Амилоидоз сердца. При вторичном А. поражение сердца встречается редко, а при первичном, в т. ч. при семейном, идиопатическом и далеко зашедшем старческом А., — у 80—100% больных. Клинически А. сердца проявляется в виде рестриктивной миокардиопатии, характеризующейся умеренным увеличением веса (не находящим иного патогенетического объяснения) и прогрессирующей сердечной недостаточностью, довольно быстро становящейся рефрактерной к лечению сердечными гликозидами. На ЭКГ отмечают сниженный вольтаж QRS-комплексов, различные нарушения внутрисердечной проводимости и другие признаки поражения миокарда. У многих больных обнаруживают протеинурию, значительно большую, чем при застойной почке. Нередко выявляется гипопротеинемия, гипоальбуминемия и гиперглобулинемия при нормальном содержании холестерина в сыворотке крови. Кроме прогрессирующей сердечной недостаточности, при А. сердца возможны (в качестве редких синдромов) констриктивный перикардит, поражение эндокарда. Иногда амилоид откладывается в стенках интрамуральных коронарных артерий, суживая их просвет; это может проявляться соответствующими изменениями миокарда на ЭКГ, в т. ч. и очагового характера.
Ряд авторов выявлял А. щитовидной железы. Внешне уплотненная и увеличенная щитовидная железа при этом напоминает тиреоидит Хасимото (см. Хасимото болезнь).
Амилоидоз желудочно-кишечного тракта. Сведения о частоте вовлечения жел.-киш. тракта в А. разноречивы. Частоту поражения жел.-киш. тракта при А. определить сложно, т. к, нек-рые симптомы связаны не с собственно А., а с теми заболеваниями, на основе к-рых он развился.
Нарушения со стороны жел.-киш. тракта (вздутие живота, понижение аппетита, дисфункция кишечника) могут быть уже в начальной стадии А. Показателем поражения капилляров кишечника является повышенное выведение из крови альбумина, меченного I131, стеаторея.
Синдром мальабсорбции при А. описывается часто и связан с отложением амилоида во внутренней оболочке кишечника (см. Мальабсорбции синдром). Поносы, возникающие в результате кишечного дисбактериоза, а также нефротического отека слизистой оболочки кишечника и уремических изменений ее, значительно ухудшают состояние больных, т. к. ведут к дальнейшему понижению питания, изменениям в водном и электролитном балансе. Нередко отмечаемые запоры связаны с развитием А. в стенке кишечника, что приводит к ее утолщению и утрате моторной функции.
Данные ректороманоскопии не дают специфической картины; гистологическое изучение биопсированной слизистой оболочки и нахождение амилоида — более ценные диагностические показатели. В литературе описан ряд случаев с резко выраженной симптоматикой поражения жел.-киш. тракта. К ним относится макроглоссия, сопровождающаяся дизартрией (см.), дисфагией (см.), затруднением акта жевания. Описаны случаи А. с признаками вовлечения пищевода, желудка, с выраженными нарушениями моторики их, с болями в «подложечной» области, тошнотой, анорексией, рвотой. Массивные отложения амилоида могут привести к обтурации просвета сосудов, местным инфарктам, изъязвлениям с кровотечением. Известно, что по частоте поражения печень является третьим органом (после селезенки и почек) при А. Существенной разницы в характере патотопографии амилоида в печени и в клинических проявлениях при различных типах А. нет. Одним из патогномоничных симптомов считается гепатомегалия. Как правило, печень бывает плотноватой с заостренным краем, безболезненной при пальпации.
Гепато- и спленомегалия могут быть связаны также и с особенностями течения тех заболеваний, при к-рых А. развился (напр., ревматоидный артрит, септический эндокардит, сердечная недостаточность). А. печени обычно не сопровождается функциональными нарушениями: казуистически редки желтуха и портальная гипертензия; не меняется активность энзимов (в т. ч. трансаминаз), проба с бромсульфалеином и сканограммы не изменены. При обратном развитии А. печень может сократиться до нормальных размеров. Пункционная биопсия печени имеет диагностические преимущества перед другими методами.
А. поджелудочной железы мало проявляется. Иногда у таких больных можно обнаружить преходящую небольшую глюкозурию и изменения активности нек-рых ферментов — трипсина и др.
Поражения нервной системы при амилоидозе. Выраженная неврологическая симптоматика свойственна отдельным формам генетически обусловленного А., когда клиническая картина напоминает тяжелую симптоматику множественного склероза и включает раннюю потерю температурной чувствительности, нарушения мочеотделения, импотенцию, прогрессирующие параличи нижних конечностей, распространяющиеся трофические расстройства. При вторичном А. поражение нервной системы обычно возникает в терминальной (уремической) стадии болезни.
Наследственный А. выделен лишь в последние годы, и до наст, времени нет цельного представления о клинической картине болезни. Имеются попытки объединить наследственные формы в группы (см. выше).
Прогноз
Прогноз зависит от характера заболевания, при к-ром развился А., осложнений, связанных как с основным заболеванием, так и с самим А. или с проводившимся лечением (применение кортикостероидов). Наличие распространенного лимфогранулематоза, иноперабельной опухоли, миеломы определяет злокачественность течения и летальный исход А. уже в ближайшее время после возникновения протеинурии.
Диагноз
Диагноз первичного системного, наследственного А. основан на тщательном генетическом анализе. Наличие заболевания у других членов семьи или в нескольких поколениях решает вопрос в пользу наследственного характера болезни. Старческий А. имеет полиморфные клинические проявления в зависимости от локализации отложений амилоида и нарушения функции органов.
Появление и прогрессирование протеинурии, возникновение нефротического синдрома или почечной недостаточности, когда сущность нефропатии остается неясной или когда имеются стойкая тяжелая сердечная недостаточность, невропатия, не объяснимые другими причинами, заставляют думать прежде всего об А.
Вероятность диагноза А. увеличивается при обнаружении гепато- и спленомегалии.
Наиболее достоверным методом диагностики А. признается биопсия органов (почек, слизистой оболочки прямой кишки, десны, лимф, узлов, печени), способствующая распознаванию и основного заболевания, при к-ром развился А. (туберкулез, злокачественная опухоль). Биопсией почек удавалось обнаружить амилоид в медуллярном слое за несколько лет до начала протеинурии, что позволило выделить преклинический период болезни и выдвинуть вопрос о своевременной профилактике амилоидоза.
Лечение
Лечение А. — это чаще всего лечение заболевания почек, выявляемого в стадии нефротического синдрома. При достаточной азотовыделительной функции почек назначается полноценный пищевой режим с ограничением поваренной соли, регулируется нарушенный баланс электролитов, осторожно применяются мочегонные и гипотензивные препараты. В период почечной недостаточности используется весь арсенал средств, применяемых при уремии. В стадии терминальной почечной недостаточности показано лечение гемодиализом. Дальнейшие перспективы лечения А. открываются с применением трансплантации почек.
Имеющиеся данные о патогенезе А. позволяют наметить следующие основные пути лечения: 1) устранение факторов, способствующих образованию амилоида; 2) торможение продукции амилоида; 3) воздействия, приводящие к рассасыванию амилоида.
Следует подчеркнуть важное значение активного лечения основного заболевания, при к-ром развился А. Это относится к вторичному А. при хрон. инфекциях и нагноительных процессах.
Удаление очага хрон. нагноения, применение больших доз антибиотиков и сульфаниламидов иногда настолько улучшают состояние больного и показатели функций почек, что можно говорить об обратном развитии А. Напр., удаление злокачественной опухоли почек приводило в течение 20 мес. к сокращению размеров амилоидной печени (диагноз был подтвержден гистологически). Активное лечение затяжного септического эндокардита вызывало рассасывание амилоида в ткани печени, констатированное при повторных биопсиях.
Возможно, что патогенетическим методом воздействия на развивающийся А. служит длительное применение сырой печени и препаратов 4-ам1шохинолинового ряда. Речь идет о дополнительном введении нек-рых аминокислот, ферментов, а синтетические противомалярийные средства, такие как хингамин (делагил), влияя на функцию ретикуло-эндотелия, способствуют торможению образования, а возможно, и резорбции уже выпавшего амилоида.
Введение кортикостероидов, цитостатиков и антплимфоцитарной сыворотки при А. не показано.
Амилоидоз кожи
А. кожи — отложение амилоидных масс в различных слоях кожи. Различают А. кожи локализованный и системный, первичный и вторичный.
Первичный локализованный амилоидоз кожи (поражена только кожа) имеет следующие разновидности:
1. Амилоидный лихен — lichen amyloidosus (Freudenthal) или amy-loidosis cutis localis nodularis et dissemuiata (Gutmann) — многочисленные, мелкие, тесно расположенные, но не сливающиеся, плотные, частично блестящие, полупросвечивающие, конические или плоские узелки цвета нормальной кожи или синюшно-фиолетовые. Локализуются на разгибательной поверхности конечностей, преимущественно в области голеней (цветн. табл., ст. 72, рис. 5). Заболевание сопровождается мучительным зудом. В результате постоянных расчесов возникает резко выраженная лихенизация и нередко рубцовая атрофия пораженной кожи («биопснрующий зуд»).
2. Пятнистый А. кожи — amyloidosis maculosa (Palitz, Peck) — одиночные или множественные, округлые или линеарные, слегка возвышающиеся розовато-коричневого цвета пятна небольших размеров, локализующиеся чаще на коже конечностей. Зуд не постоянен.
3. Опухолевидный А. кожи — опухолевидные образования различных размеров, единичные или множественные, располагаются в дерме и гиподерме, чаще в области лица, груди, гениталий. Кожа над ними атрофична, легко собирается в складки, напоминает папиросную бумагу; на ней могут образовываться пузыри с вялой покрышкой и прозрачным содержимым.
Болеют первичным локализованным А. кожи чаще пожилые мужчины, известны случаи заболевания в нескольких поколениях.
Вторичный локализованный амилоидоз кожи — отложение амилоида в коже, пораженной до этого другими дерматозами (себорейные бородавки, кератозы, язвы трофические и пиодермические, Боуэна болезнь и др.).
Первичный системный амилоидов кожи — amyloidosis cutis metabolica (Königstein) — отложение в коже амилоида при общем А. с поражением внутренних органов (см. выше). Характеризуется появлением на бледной, фарфороподобной коже разнообразных высыпаний: а) гладких восковидных мелких папул, цвета кожи или желтоватых, на лице, особенно в области глазниц, шее и гениталиях (цветн. табл.), нередко с геморрагическим компонентом; б) гладких плотных желтоватых бляшек различных размеров, чаще на коже конечностей; в) узлов и опухолевидных образований с преимущественным расположением на туловище; г) пурпуры (петехии, экхимозы), как правило, на лице, шее, слизистых оболочках. Кроме того, могут наблюдаться алопеция, обычно универсальная; дистрофия ногтей (ногтевые пластины становятся тусклыми, ломкими, возможна анонихия); белый акантоз (Сорро) — бородавчатые разрастания и утолщение кожи в складках, подобные acanthosis nigricans (см.), но гипохромные; эритродермия желтовато-красного оттенка [«оранжевый человек» (Gougerot, Grupper)], макрохейлия, макроглоссия, ксеростомия. Зуд обычно отсутствует. Вторичный системный А. кожи встречается крайне редко.
Гистопатология. Амилоидные массы могут откладываться в сосочковом слое дермы, непосредственно под эпидермисом (амилоидный лихен, пятнистый амилоидоз кожи) или располагаться диффузно в дерме и гиподерме, вокруг сальных желез и волосяных сумок, поражая глубокие сосуды дермы по периколлагеновому типу (см. выше Первичный системный амилоидоз кожи).
Для подтверждения диагноза А. кожи используют пробу с конго красным (Маркионини — Джона), для чего в пораженную кожу вводят 1,5% раствор конго красного (1 мл подкожно или 0,1 мл внутрикожно). Участки с отложением амилоида через 24—48 час. отчетливо и стойко окрашиваются в красный цвет, а окружающая кожа окрашивается очень слабо.
Течение А. кожи хроническое. Прогноз при локализованных формах благоприятный, при системном А. кожи — зависит от степени поражения внутренних органов. Дифференцировать А. кожи следует с нейродермитом, красным плоским лишаем, склеродермией.
Лечение нередко безрезультатно. Рекомендуются концентрат витамина А, антиметаболиты, диатермокоагуляция, рентгенотерапия. Наружно назначают желатино-цинковые повязки.
Амилоидоз у детей
Общим А. дети болеют редко, у новорожденных он почти не встречается. У детей дошкольного и школьного возраста А. развивается как осложнение (вторичный А.) и реже вне связи с каким-либо заболеванием (первичный).
| Заболевание | Возраст | Анамнез | Клинические проявления | Течение | Данные лабораторных исследований |
|---|---|---|---|---|---|
| Амилоидоз с нефротическим синдромом | Дошкольный, школьный | Хронические нагноительные процессы, хронические инфекции, напр. туберкулез, ревматоидный артрит — болезнь Стилла и др. | Появление первых признаков амилоидоза через один — три года от начала основного заболевания. Постепенное развитие отеков. Увеличенные печень и селезенка — гладкие, плотные. В финале заболевания геморрагический синдром | Длительное, прогрессирующее | В начале заболевания транзиторная протеинурия, затем массивная; α2-глобулинемия, гипоальбуминемия, редко гиперхолестеринемия. Ускоренная РОЭ, лейкоцитоз, тромбоцитоз. Раннее нарушение концентрационной способности почек. Положительная проба с конго красным. При пункционной биопсии почек наблюдается отложение амилоида |
| Врожденный липоидный нефроз | Первые месяцы жизни или 1—2 года | Указание на семейный характер заболевания. Заболевание почек у матери | Постепенное развитие отеков | Волнообразное течение | Массивная протеинурия. гиперхолестеринемия. Отсутствие биохимических и лабораторных признаков активности процесса. При пункционной биопсии почек наблюдаются микрокистозные изменения почечной ткани |
| Диффузный гломерулонефрит (нефротическая форма) | Чаще в дошкольном | Развивается через 1—3 недели после ангины, обострения тонзиллита, скарлатины, острого респираторного заболевания | Быстрое развитие периферических и полостных отеков, через 1—3 недели после перенесенной инфекции увеличение печени | Острое или волнообразное | Выраженная протеинурия, гипопротеинемия, α-глобулинемия, гиперхолестеринемия |
| Цирроз печени | В любом возрасте | Развивается чаще после перенесенной болезни Боткина | Сухость кожных покровов, расчесы, дистрофия. Печень каменистой плотности, край неровный, часто увеличенная селезенка. Выраженная венозная сеть на животе. Вначале появляется асцит, потом отеки на верхней половине туловища | Медленное | Ускоренная РОЭ. При пункционной биопсии почек данные за мембранозный и мембранозно-пролиферативный нефрит |
В моче уробилин, незначительная протеинурия. Анемия, лейкопения, тромбоцитопения. Повышение альдолазы, трансаминазы, измененные осадочные пробы. Гипоальбумияемия, гипергаммаглобулинемия. Замедленное выведение бром-сульфалеина. При пункционной биопсии печени отсутствие отложения амилоида
Чаще А. у детей встречается при ревматоидном артрите, значительно реже при остеомиелите, туберкулезе, системной красной волчанке, злокачественных опухолях, лимфогранулематозе, периодической болезни.
Частота А. у детей, больных ревматоидным артритом, колеблется от 2,7 до 15,6% , составляя в среднем 5%.
М.П. Матвеев и соавт. выявили А. у детей, больных нефритом с выраженным нефротическим синдромом (3,7% случаев).
По данным Б. М. Кавалива, среди детей с костно-суставным туберкулезом амилоидный нефроз был отмечен в 10 раз реже, чем у взрослых (примерно в 0,2%).
А. у детей протекает тяжело, прогрессирует быстрее, чем у взрослых. У больных с наименьшей продолжительностью основного заболевания наблюдается изолированное поражение почек, селезенки или печени.
У детей в основном поражаются почки. Появляется протеинурия без патологических элементов в осадке. Вначале она непостоянна и незначительна, при гломерулонефрите — стойкая. Позднее протеинурия имеет тенденцию к нарастанию, причем осадок мочи скудный, с микрогематурией, микролейкоцитурией и единичными гиалиновыми и зернистыми цилиндрами. Постепенно нарастает бледность кожных покровов, появляется гепатомегалия, спленомегалия, держится ускоренная РОЭ, тромбоцитоз. Наблюдается стойкая диспротеинемия за счет увеличения α2-глобулинов. По мере прогрессирования А. нарастают отеки, протеинурия, присоединяется полиурия, никтурия, гипостенурия. Возрастает α-глобулинемия, в меньшей степени — β-глобулинемия. Усиливается гипопротеинемия, гипоальбуминемия. У большинства детей определяется гипер-липидемия, реже — гиперхолестеролемия. В крови ускоренная РОЭ, нейтрофильный лейкоцитоз, анемия. В дальнейшем нарастает азотемия, появляется геморрагический синдром, развивается уремия.
При ревматоидном артрите по мере прогрессирования А., нарастания гуморальных показателей активности процесса экссудативные явления в суставах стихают. При периодической болезни часто встречается гипер-тензия и реже гепатолиенальный синдром. При других коллагеновых заболеваниях А. обычно не сопровождается нефротическим синдромом или он бывает слабо выражен.
Для ранней диагностики А. имеет большое значение электрофоретическое исследование белков сыворотки крови и биопсия органов. Проба с конго красным мало чувствительна, однако для дифференциальной диагностики ее следует применять.
Дифференциальная диагностика А. у детей — см. таблицу на стр. 359.
При амилоидозе проводится лечение основного патологического процесса, лежащего в основе его развития. Рано начатое лечение и ликвидация основного заболевания могут привести к обратному развитию А.
При развитии нефротического синдрома применяют лечение в соответствии с клиническими симптомами.
В последнее время для лечения А. используют препараты 4-аминохинолинового ряда (хлорохин, резохин, делагил и др.). Спорным остается вопрос о применении стероидных гормонов. По мнению большинства авторов, кортикостероидные препараты при лечении А. мало эффективны.
Библиогр.: Андреева Н. Е. и Алексеев Г. А. Амилоидоз при миеломной болезни (параамилоидоз), Пробл. гематол. и перелив, крови, т. 13, № 3, с. 16, 1968; Виноградова О. М. и др. Первичный семейный амилоидоз, Тер. арх., т. 41, № 2, с. 105, 1969, библиогр.; Давыдовский И. В. Огнестрельная рана человека, т. 2, с. 351, М., 1954; Рукосуев В. С. Иммуноморфологическая идентификация фибрина в амилоидных массах, Арх. патол., т. 27, № 9, с. 32, 1965; Серов В. В. Некоторые спорные вопросы классификации амилоидоза, там же, т. 32, № 6, с. 8, 1970, библиогр.; Тареев Е. М. и др. К проблеме нефротического синдрома, Тер. арх., т. 35, № 11, с. 9, 1963, библиогр.; Andrade С. а. о. Hereditary amyloidosis, Arthr. and Rheum., v. 13, p. 902, 1970, bibliogr,; Вriggs G. W. Amyloidosis, Ann. intern. Med., v.55, p. 943, 1961, bibliogr.; Cathcart E. S., Skinner M. a. Cohen A. S. Immunogenicity of amyloid, Immunology, v. 20, p. 945, 1971, bibliogr.; Соhen A. S. а. Сalkins L. E. Electron microscopic observations on a fibrous component in amyloid of diverse origins, Nature (Lond.), v. 183, p. 1202, 1959; Cohen A. S., Gross E. a. Shirahama T. The light and electron microscopic autoradiographic demonstration of local amyloid formation in spleen explants, Amer. J. Path., v. 47, p. 1079, 1965, bibliogr.; Druet R. L. a. Janigan D. T. Rates of induction, lymphocyte depletion and thymic atrophy, ibid., v. 49, p. 911, 1966, bibliogr.; Gafni J., Sonar E. a. Heller H. The inherited amyloidoses, Lancet, v. 1, p. 71, 1964; Glenner G. G. a. o. Amyloid, J. Histochem. Cytochem., v. 16, p. 633, 1968, bibliogr.; Heller H., Sohar E. a. Gafni J. Classification of amyloidosis with special regard to the genetic types. Path, et Microbiol. (Basel), v. 27, p. 833, 1964, bibliogr.; Heller H. a. o. Amyloidosis, J. Path. Bact., v. 88, p. 15, 1964, bibliogr.; Кilpatriсk Т. Р., Horack H. M. a. Moore C. B. «Stiff heart» syndrome, Med. clin. N. Amer., v. 51, p. 959, 1967, bibliogr.; Schmitz-Moormann P. Zur Biochemie des Amyloid, Virchows Arch. path. Anat., Bd 339, S. 45, 1965; Schwartz Ph. Amyloidosis, Springfield, 1970, bibliogr.; Senn H. I. u. a. Zur klinischen Diagnose der Amyloidose, Schweiz. med. Wschr., S. 1363, 1966, Bibliogr.; Shirahama T. a. Cohen A. S. Highresolution electron microscopic analysis of the amyloid fibril, J. cell. Biol., v. 33, p. 679, 1967, bibliogr.; Stiller D. u. Кatenkanp D. Zur Pathogenese dcr senilen Amyloidose, Virchows Arch. path. Anat., Bd 352, S. 209, 1971; Vazquez J. J. a. Dixоn F. J. Immunohistochemical analysis of amyloid by the fluorescence technique, J. exp. Med., v. 104, p. 727, 1956.
А. кожи — Левер У. Ф. Гистопатология кожи, пер. с англ., с. 291, М., 1958; Вanerjее В. N. a. Duttа А. К. Lichen amyloidosis, Int. J. Derm., v. 9, p. 290, 1970, bibliogr.; Brownstein M. H. a. Helwig Е. В. Cutaneous amyloidoses, Arch. Derm., v. 102, p. 8, 1970, bibliogr.; Potter B. S. a. Johnson W. C. Primary localized amyloidosis cutis, ibid., v. 103, p. 448, 1971, bibliogr.
A. у детей — Думнова А. Г. К клинике амилоидоза почек при инфекционном неспецифическом (ревматоидном) артрите у детей, Педиатрия, № 8, с. 47, 1968, библиогр.; Матвеев М. П. и др. О нефротическом синдроме при амилоидозе у детей, Урол. и нефрол., № 5, с. 23, 1971; Щерба М. Л. Общий амилоидов, Л., 1957, библиогр.; Яковлева А. А. и Брязгунов И. П. Амилоидов у детей, Педиатрия, № 6, с. 81, 1971, библиогр.; Ansell В. М. а. Вуwaters E. G. L. Rheumatoid arthritis (Still's disease), Pediat. Clin. N. Amer., v. 10, p. 921, 1963, bibliogr.; Strauss R. G., Schubert W. K. а. Мс Adams A. I. Amyloidosis in childhood, J. Pediat., v. 74, p. 272, 1969, bibliogr.
Источники:
- Большая медицинская энциклопедия. Том 1/Главный редактор академик Б. В. Петровский; издательство «Советская энциклопедия»; Москва, 1974.- 576 с.
|
ПОИСК:
|
При копировании материалов проекта обязательно ставить активную ссылку на страницу источник:
http://sohmet.ru/ 'Sohmet.ru: Библиотека по медицине'