АЗОТИСТЫЙ ОБМЕН
Расстановка ударений: АЗО`ТИСТЫЙ ОБМЕ`Н
Содержание:
Переваривание белков и других азотсодержащих веществ Тканевой обмен аминокислот Участие аминокислот в процессах биосинтеза Участие аминокислот в процессах катаболизма Образование конечных продуктов обмена простых белков Тканевой обмен нуклеотидов Синтез ДНК и РНК Катаболизм ДНК и РНК Регуляция процессов азотистого обмена Радиоизотопное исследование азотистого обмена Патология азотистого обмена Азотистый обмен в облученном организме Изменение азотистого обмена в процессе старения Особенности азотистого обмена у детей
Азотистый обмен — совокупность химических превращений азотсодержащих веществ в организме.
А. о. включает обмен простых и сложных белков, нуклеиновых кислот, продуктов их распада (пептидов, аминокислот и нуклеотидов), содержащих азот жироподобных веществ (липидов), аминосахаров, гормонов, витаминов и др.
Для нормального течения процессов жизнедеятельности организм должен быть обеспечен необходимым количеством усвояемого азота. Главнейшей составной частью и основным источником азота пищи человека являются белковые вещества (см. Белки).
Суточная норма белка в питании взрослого человека, принятая в СССР, составляет 100 г белка или 16 г белкового азота при трате энергии в 2500 ккал. Однако и значительно меньшие количества белка могут обеспечить азотистое равновесие, т. е. состояние, при к-ром количества вводимого и выводимого азота одинаковы. После приема белковой пищи основной обмен (см. Обмен веществ и энергии) повышается больше, чем это обусловлено калорийной ценностью белка. Это явление получило наименование «специфически динамическое действие» белковой пищи. Механизм этого явления не вполне ясен. По-видимому, нек-рые аминокислоты — продукты расщепления белка — участвуют в реакциях, связанных с гидролизом АТФ и образованием АДФ, обусловливая повышенное потребление кислорода.
При общем голодании или при недостаточном азотистом питании количество выводимого с мочой и калом азота превышает количество вводимого с пищей — состояние отрицательного азотистого баланса. В том случае, когда количество вводимого азота превышает количество выводимого, наступает состояние положительного азотистого баланса, что характерно для растущего организма, при процессах регенерации и т. д.
При составлении или оценке рациона питания необходимо учитывать полноценность белков, характеризующуюся содержанием в них незаменимых аминокислот, т. е. таких, к-рые не могут образоваться из других соединений в организме (см. Аминокислоты).
Суточная потребность организма человека в различных незаменимых аминокислотах неодинакова (табл. 1).
| Аминокислота | Количество, гарантирующее азотистое равновесие | Минимальное количество, обеспечивающее азотистое равновесие |
|---|---|---|
| L-триптофан | 0,5 | 0,25 |
| L-фенилаланин | 2,2 | 1,1 |
| L-лизин | 1,6 | 0,8 |
| L-треонин | 1,0 | 0,5 |
| L-метионин | 2,2 | 1,1 |
| L-лейцин | 2,2 | 1,1 |
| L-изолейцин | 1,4 | 0,7 |
| L-валин | 1,6 | 0,8 |
Переваривание белков и других азотсодержащих веществ
Для высокоорганизованных позвоночных животных, в т. ч. и для человека, началом процессов А. о. следует считать переваривание в жел.-киш. тракте простых и сложных белков, а также других сложных азотистых соединений с последующим всасыванием продуктов их расщепления.
Переваривание белков начинается в желудке под влиянием ферментов пепсина (см.) и гастриксина, вырабатываемых в слизистой оболочке желудка в неактивной форме — в виде зимогенов (проферментов).
Кислая среда, необходимая для активации зимогенов, обеспечивается соляной к-той, секретируемой железами слизистой оболочки дна желудка (обкладочные клетки). Пепсин (оптимум рН ок. 2) и гастриксин (оптимум рН 3—4) представляют собой протеазы — эндопептидазы, к-рые разрывают пептидные связи между аминокислотами, расположенными внутри пептидных цепей молекулы белка (см. Пептидгидролазы).
При накоплении в желудке пищевой массы, обладающей достаточно кислой реакцией, рефлекторно раскрывается пилорический жом, и пищевая масса порциями поступает в двенадцатиперстную кишку, а затем в нижележащие отделы тонкой кишки, где в дальнейшем расщеплении пептидных связей участвуют ферменты сока поджелудочной железы — трипсин (см.), химотрипсин (см.) и карбоксипептидаза (см.) и ферменты кишечника — амино- и дипептидазы.
Трипсин и химотрипсин относятся к эндопептидазам (оптимум рН ок. 8,0), карбокси- и аминопептидазы — к экзопептидазам; они расщепляют крайнюю пептидную связь соответственно со стороны свободной карбоксильной и аминогруппы. При образовании дипептидов они расщепляются дипептидазами. Параллельно перевариванию простых белков в тонкой кишке происходит расщепление нуклеопротеидов, а также дезоксирибонуклеиновых (ДНК) и рибонуклеиновых (РНК) кислот.
В результате последовательного проявления гидролитической активности ферментов пищеварительных желез, а также микроорганизмов кишечника простые и сложные белки, а также другие биополимеры распадаются, и продукты распада (низкомолекулярные пептиды, аминокислоты, нуклеотиды, нуклеозиды) всасываются в тонком кишечнике и поступают в кровь.
Параллельно происходит ферментативный распад тканевых белков под влиянием тканевых протеаз — катепсинов (см.) и пептидаз (см. Пептидгидролазы). Образующиеся ори этом продукты распада также попадают в кровь и разносятся во все органы и ткани.
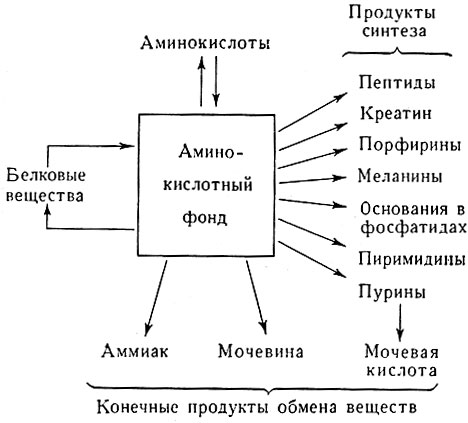
Рис. 1. Схема использования аминокислотного фонда в организме
Тканевой обмен аминокислот
Фонд аминокислот, образовавшийся в результате ферментативного расщепления пищевых продуктов или продуктов распада тканей, расходуется на биосинтез белков и многих других соединений, свойственных только данному организму, на энергетические затраты, а также на образование конечных продуктов азотистого обмена, подлежащих выведению (рис. 1).
Участие аминокислот в процессах биосинтеза
Синтез специфических для данного организма белков находится под контролем молекул ДНК, входящих в состав хроматина клеточных ядер.
На одном из тяжей ДНК (в месте ее раскручивания) по закону комплементарности (см. Генетический код) происходит сборка (синтез) информационных, или матричных, РНК (мРНК).
К фиксированным на рибосомах мРНК подходят транспортные рибонуклеиновые кислоты (тРНК), несущие на себе предварительно активированные аминокислоты, к-рые фиксируются на мРНК. Рядом располагаются такие аминокислоты, к-рые в синтезируемом белке должны быть соединены пептидной связью, чем обеспечивается специфическая первичная структура белков со строго определенным порядком следующих друг за другом аминокислот.
В свою очередь первичная структура предопределяет, если не полностью, то в значительной мере, пространственную конфигурацию, или третичную структуру, белков, в т. ч. и белков-ферментов.
Выпадение или нарушение какого-либо звена в сложном процессе биосинтеза фермента, осуществляющего определенную реакцию в обмене веществ, может привести к тяжелым патологическим нарушениям. Так, причиной многих наследственных болезней (см.) является выпадение синтеза всего лишь одного белка-фермента (напр., гидроксилазы при фенилпировиноградной олигофрении); «ошибка» в первичной структуре У α- или β-цепей гемоглобина, заключающаяся в замене всего лишь одной из 287 аминокислот, приводит к образованию патологических форм гемоглобина с нарушенной функцией присоединения и отдачи кислорода.
Фонд аминокислот используется также при синтезе других соединений.
Напр., биосинтез пуриновых нуклеотпдов (см. Пуриновые основания), начинающийся с рибозил-5-фосфата, проходит через многочисленные стадии и завершается образованием инозиновой к-ты (инозиновая к-та затем может подвергаться превращениям в адениловую и гуаниловую кислоты). При этом требуется участие глутамина (амида глутаминовой к-ты) в качестве источника азота в 3-м и 9-м положениях, глицина — в 7-м положении и углерода — в 4-м и 5-м положениях. Аспарагиновая к-та — источник азота в 1-м положении:
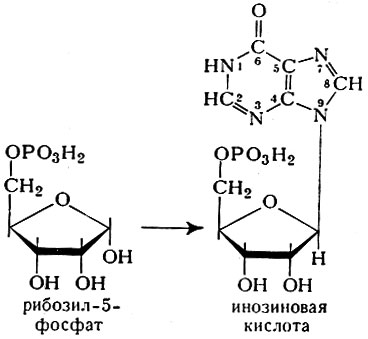
Атомы углерода (2-й и 8-й) доставляет формильное производное тетрагидрофолиевой к-ты, и, наконец, углерод на 6-м месте кольца пурина берется из бикарбоната. Эти данные представлены на схеме:
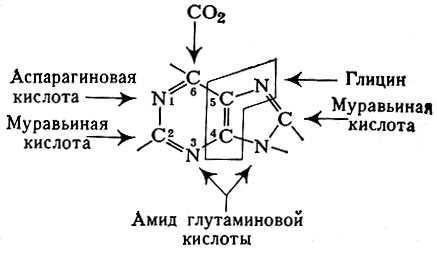
При последующем образовании адениловой к-ты (см. Аденозинфосфорные кислоты) вновь вовлекается аспарагиновая к-та, азот к-рой обеспечивает аминогруппу, стоящую при 6-м углеродном атоме пуринового кольца. При синтезе гуаниловой кислоты (см.) аминогруппа при 2-м углеродном атоме берется из глутамина.
Синтез пиримидинов начинается с образования богатого энергией соединения — карбамилфосфата:
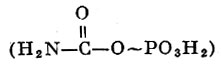
из аммиака (NH3), бикарбоната (НСО3-), аденозинтрифосфата (АТФ) как источника энергии и, наконец, N-ацетилглутаминовой к-ты в качестве активатора:
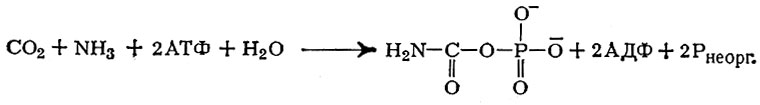
Карбамильная группа карбамилфосфата ферментативно переносится на аспарагиновую к-ту. Через образовавшуюся карбамиласпарагиновую к-ту, дигидрооротовую и оротовую кислоты (рис. 2) образуется оротидиловая к-та, переходящая в уридиловую к-ту и уридинтрифосфат (УТФ). Путем аминирования УТФ образуется цитидинтрифосфат (ЦТФ), причем эта последняя реакция представляет собой регулируемый процесс по закону обратной связи: ЦТФ тормозит образование карбамиласпарагиновой к-ты, а АТФ снимает это торможение. Т. о., образование пиримидиновых нуклеотидов, входящих в состав нуклеиновых кислот, регулируется соотношением содержания ЦТФ и АТФ.
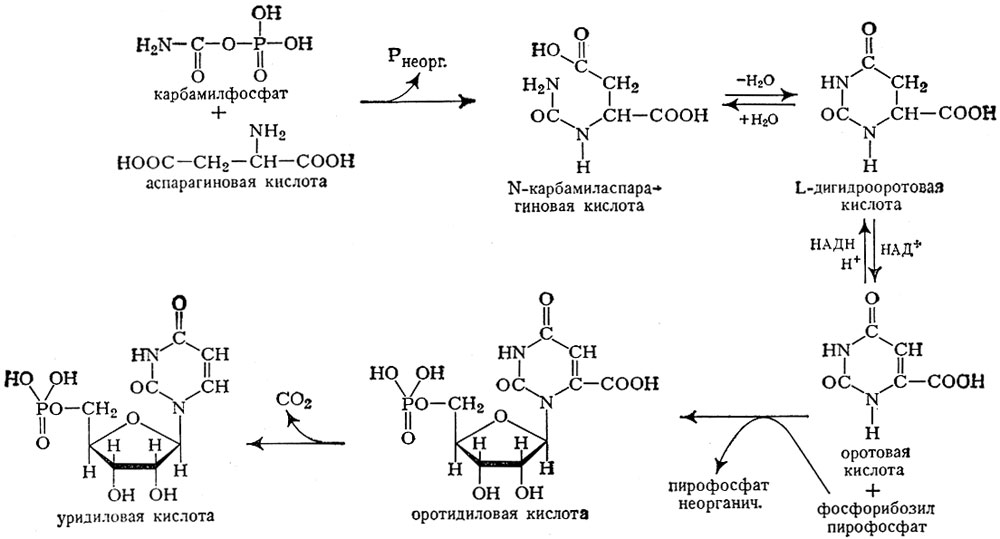
Рис. 2. Схема биосинтеза пиримидиновых оснований
Помимо образования пуриновых и пиримидиновых нуклеотидов, аминокислоты участвуют в образовании многих других физиологически важных соединений.
1. Из триптофана (α-амино-β-индолпропи оновой к-ты):
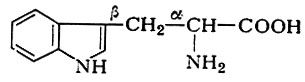
в результате ряда последовательных ферментативных превращений образуется никотиновая к-та, выполняющая функцию антипеллагрического витамина и участвующая в виде никотинамида
в биосинтезе никотпнамидных коферментов НАД и НАДФ.
2. Простейшая аминокислота глицин (CH2NH2COOH), помимо участия в образовании пуринов, обеспечивает весь азот и ряд атомов углерода при биосинтезе порфиринов, составляющих структурную основу желчных пигментов и небелковой части (простетической группы) железосодержащих хромопротеидов (см.).
Глицин выполняет также роль акцептора амндиновой группы аргинина при синтезе гуанидинуксусной к-ты, N-метилпронзводное к-рой — креатин (см.)
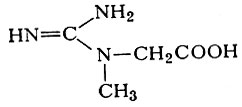
является важной составной -частью скелетной мускулатуры, сердца и мозга, а в виде фосфорилированного продукта (фосфокреатина) обеспечивает резерв богатых энергией фосфорных соединений, необходимых для функциональной активности ткани.
3. Серин
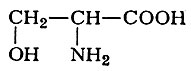
участвует в образовании сложного аминоспирта — сфингозина (см. Сфингозины), входящего в состав сфингомиелина (см. Сфинголипиды) — липида, особенно богато представленного в составе мозга и нервной ткани.
Серии участвует также в синтезе кофермента (см.) ацетилирования (КоА), ацилпроизводные к-рого представляют активную форму жирных кислот (см. Жировой обмен), участвующих в различных процессах биосинтеза и окислительного распада.
| Аминокислота | Вещество, предшественником которого являются аминокислоты |
|---|---|
| Аргинин | Спермин, спермидин, путресцин |
| Гистидин | Гистамин, эрготионеин |
| Лизин | Кадаверин, анабазин, кониин |
| Тирозин | Адреналин, норадреналин, меланин, тироксин, мескалин, тирамин |
| Триптофан | Серотонин, индол, скатол |
В табл. 2 представлены дополнительные сведения об отдельных аминокислотах, являющихся предшественниками нек-рых других биологически важных азотистых соединений.
Функциональные группы аминокислот широко вовлекаются в различные реакции обмена веществ.
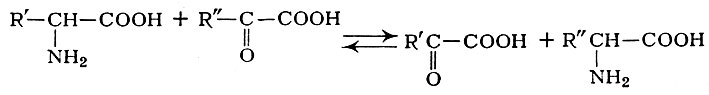
Прежде всего это относится к аминогруппам, участвующим в реакции переаминирования (см.). Эта реакция, представляющая важнейший путь ферментативного превращения аминокислот, была открыта советскими биохимиками А. Е. Браунштейном и М. Г. Крицман в 1937 г. Она заключается в обратимом ферментативном переносе α-аминогруппы α-аминокислоты на α-углеродный атом α-кетокислоты без промежуточного освобождения аммиака.
В реакциях переаминирования, катализируемых различными трансаминазами, могут участвовать не только аминогруппы α-аминокислот, но и аминогруппы аминов и ω-аминокислот (напр., β-аланина, γ-аминомасляной к-ты); акцептировать аминогруппы могут не только α-кетокислоты, но и альдегиды (напр., малоновый или янтарный полуальдегиды).
Общая схема реакции переаминирования обычно изображается в следующем виде:
Непременным участником обратимой реакции ферментативного переаминирования, выполняющим коферментную функцию, является пиридоксальфосфат (I), а также пиридоксаминфосфат (II), оба — производные витамина В6 (пиридоксина).
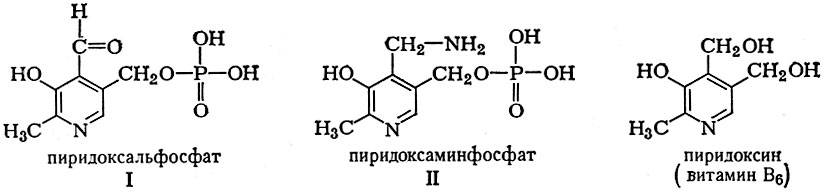
Пиридоксальфосфат принимает на себя аминогруппу аминокислоты и через стадии образования шиффовых оснований превращается в пиридоксаминфосфат (II), к-рый передает аминогруппу также через промежуточные стадии на кетокислоту, возвращаясь в первоначальное состояние (I).
Дикарбоновые аминокислоты — глутаминовая и аспарагиновая — наиболее активные участники процесса переаминирования. Под влиянием фермента глутаматдегидрогеназы осуществляется образование глутаминовой к-ты из аммиака и кетоглутаровой к-ты. Аминогруппа глутамиыовой к-ты широко транспортируется при участии аминофераз на различные α-кетокислоты и альдегиды, образуя новые аминокислоты и амины. Этим косвенным путем азот аммиака вовлекается в состав многочисленных азотистых органических веществ.
В биосинтезе ряда биологически активных соединений значительная роль принадлежит процессу метилирования. Перенос метильной группы, как правило, осуществляется аминокислотой — метионином в виде аденозилметионина, превращающегося после отдачи метильной группы в S-аденозилгомоцистеин (рис. 3).
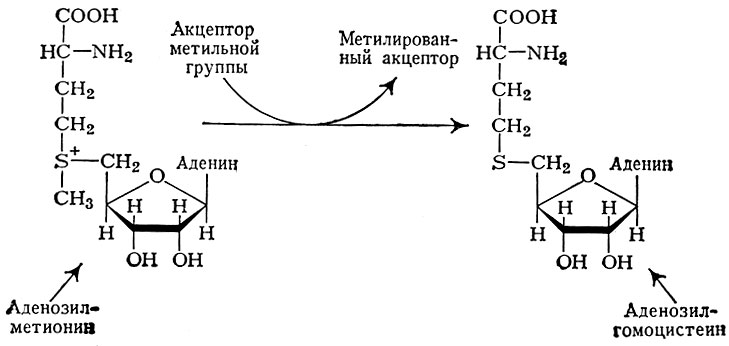
Рис. 3. Схема процесса метилирования
Акцепторы метильной группы разнообразны; к ним относятся: липиды, транспортные нуклеиновые кислоты, содержащие минорные (редкие) компоненты — метилированные нуклеотиды, гуанидинуксусную к-ту, никотинамид и др. Донорами метильных групп могут быть, помимо аденозилметионина, еще и холин, бетаины, N5-метилтетрагидрофолиевая к-та и др. (см. Метилирование).
Участие аминокислот в процессах катаболизма
Одним из путей катаболизма (деградации) аминокислот является их ферментативное декарбоксилирование (см.), приводящее к освобождению углекислого газа и образованию биогенных аминов, обладающих высокой биологической активностью, напр, гистамина
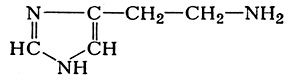
из гистидина; серотонина
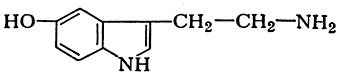
из окситриптофана; γ-аминомасляной к-ты из глутаминовой к-ты:
тирамина из тирозина
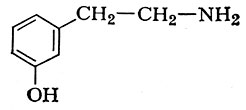
Декарбоксилирование аминокислот катализируется декарбоксилазами (см.), коферментом к-рых обычно является пиридоксальфосфат, однако механизм декарбоксилирования аминокислот остается недостаточно выясненным. В декарбоксилазе гистидина коферментная функция принадлежит остатку пировиноградной к-ты, карбоксильная группа к-рой соединена с пептидной цепью белка-фермента кислотноамидной связью:
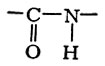
Образовавшиеся при декарбоксилировании аминокислот амины служат субстратами окисления для моноамино- и диаминооксидаз — ферментов, отличающихся друг от друга не только белковой частью, но и коферментами: митохондриалыше моноаминооксидазы (см.) принадлежат к флавопротеидам, их кофсрмент — флавинадениндинуклеотид. У диаминооксидаз коферментом служит пиридоксальфосфат. Образующиеся при дезаминированип монаминов аммиак и альдегиды претерпевают дальнейшие превращения: обезвреживание аммиака происходит преимущественно путем образования мочевины (см.), углеродный скелет аминов (в виде альдегидов) подвергается дальнейшему окислению.
Другим процессом деградации L-аминокислот является их окислительное дезаминирование (см.), идущее с образованием аммиака и кетокислот. Эта реакция протекает в организме высших животных и человека очень медленно (в противоположность окислительному образованию аммиака из D-аминокислот), однако может осуществляться быстрее косвенным путем: сначала при персаминировании образуется α-глутаминовая к-та, к-рая затем при дезаминировании является источником кетоглутаровой к-ты и аммиака. Следует, однако, учесть, что в реакции дезаминирования равновесие смещено в сторону восстановительного образования глутаминовой к-ты, т. е. слева направо:

Пути образования аммиака из аминокислот остаются недостаточно ясными.
В последнее время Г. X. Бунатяном и его сотрудниками большое значение в процессе образования аммиака (в частности, в ц. н. с. и в печени) приписывается отщеплению NH2-группы аденина, находящегося в составе никотинамидадениндпнуклеотида (НАД). Продуктом этой реакции является дезамгшоникотинамид-адениндинуклеотид (деНАД):
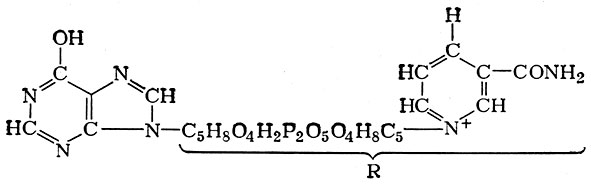
Дезаминоникотинамидадениндинуклеотид (деНАД)
Последующее аминирование деНАД осуществляется при участии аспа-рагиновой к-ты, протекает с образованием промежуточного продукта (НАД-янтарной к-ты) и после отщепления фумаровой к-ты приводит к восстановлению первоначальной структуры НАД (рис. 4):
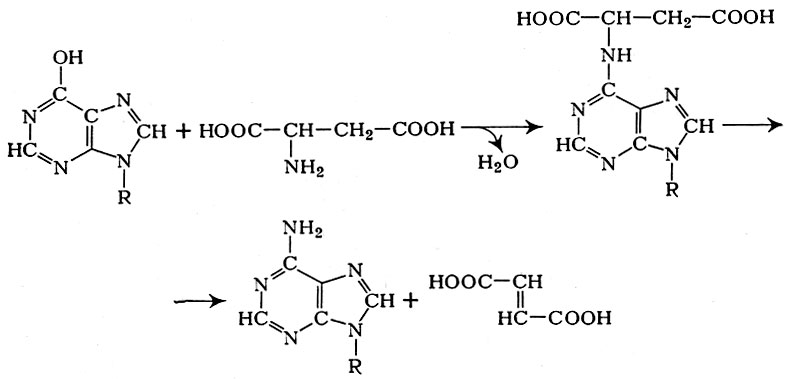
Рис. 4. Схема превращении дезамнноникотинамидадениндннуклеотида
Следовательно, по Г. X. Бунатяну, дезаминирование α-аминокислот с образованием аммиака протекает через образование аспарагиновой к-ты путем переаминирования, передачи аминогруппы на деНАД, образования НАД и отщепления аммиака от НАД.
В наст. время еще невозможно оценить, насколько широко этот процесс представлен в организме и каково его биологическое значение. ДеНАД приписывают высокую биологическую активность в качестве фактора, легко проникающего как в окисленной, так и в восстановленной форме через мембрану митохондрий и значительно повышающего энергетическую эффективность окислительного фосфорилирования.
Образование конечных продуктов обмена простых белков
Возникший в процессах обмена веществ аммиак и безазотистый остаток аминокислот претерпевают своеобразные превращения. Основной путь нейтрализации и связывания аммиака у уреотелических животных заключается в синтезе мочевины, протекающем в печени и состоящем из серии последовательных ферментативных реакций. Первый этап этого процесса заключается в образовании карбамилфосфата (так же, как при синтезе пиримидиновых оснований), затем карбамильная группа акцептируется орнитином.
Образовавшийся при этом цитруллин эндергонически реагирует с аспарагиновой к-той. Образовавшаяся аргининянтарная к-та подвергается расщеплению: один из продуктов реакции — фумаровая к-та — включается в цикл трикарбоновых кислот, другой — аргинин — гидролитически расщепляется аргиназой на мочевину и орнитин (рис. 5). Последний вновь включается в цепь превращений, приводящих к образованию мочевины. Процесс этот, получивший наименование орнитинового цикла, протекает в печени, хотя отдельные его реакции представлены также в сердце, ткани мозга и др.
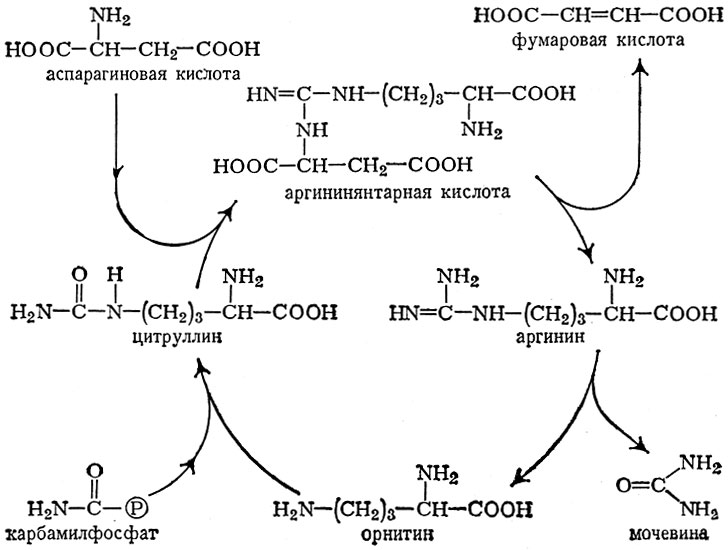
Рис. 5. Орнитиновый цикл
Т. о., азот, выводимый из организма в виде мочевины, наполовину берется из аммиака и наполовину из аспарагиновой к-ты.
Нейтрализация образующегося в организме аммиака происходит еще путем синтеза амидов — аспарагина и глутамина. Амидная группа последнего участвует в синтезе пуринов, нуклеиновых кислот и т. д.
Нейтрализация аммиака у урикотелических животных (рептилии, птицы) связана с образованием мочевой кислоты (см.).
Безазотистая часть аминокислот, как правило, включается через многочисленные промежуточные этапы в разные стадии окислительных превращений по циклу трикарбоновых кислот (см. Трикарбоновых кислот цикл).
Согласно схеме, приведенной на рис. 6, отчетливо выявляется роль аминокислот в обеспечении энергетических запросов организма. Нарушения в превращениях тех или иных аминокислот часто генетически обусловлены и являются причиной различных болезней.
Причиной нарушений, как правило, является дефект в одном специфически действующем ферменте или в серии ферментативных реакций. Эти нарушения могут возникнуть, напр., в силу недостаточного образования или слишком быстрого расщепления кофермента, участвующего во многих ферментативных процессах.
Тканевой обмен нуклеотидов
Продукты распада нуклеопротеидов и нуклеиновых кислот — нуклеотиды и нуклеозиды — претерпевают в органах п тканях различные превращения.
Синтез ДНК и РНК
Нуклеотиды — как пуриновые, так и пиримидиновые — участвуют в синтезе нуклеиновых кислот в клеточных ядрах. Синтез ДНК осуществляется ферментами — ДНК-полимеразами, для которых субстратами служат дезоксирибонуклеозидтрифосфаты.
Синтез ДНК сопровождается освобождением молекул пирофосфата в количестве, соответствующем числу молекул нуклеозидтрифосфатов, вступивших в реакцию. ДНК (образец) и вновь синтезированный полинуклеотид образуют вместе двутяжную ДНК. Схема этого процесса может быть представлена в следующем виде:
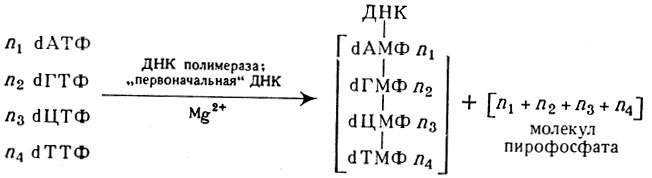
Схема биосинтеза ДНК
Буква «d» перед символом нуклео-зидтрифосфата или мононуклеотидов в синтезированной молекуле ДНК обозначает, что в биосинтезе участвуют нуклеотиды, в к-рых пентоза представлена дезоксирибозой, т. е. дезоксирибонуклеотиды. Образование дезоксирибонуклеотидов происходит в результате сложного процесса восстановления рибонуклеотидов при действии нечувствительного к нагреванию белка — тиоредоксина.
Восстановленная форма тиоредоксина образуется под действием редуктазы (фермента флавопротешговой природы), коферментом к-рого служит восстановленный никотинамидадениндпнуклеотидфосфат (НАДФ) по схеме:

Образовавшаяся восстановленная форма тпоредоксина участвует в образовании дезоксинуклеотиддифосфатов (дНДФ) путем переноса редуцирующих эквивалентов на акцептирующие пх нуклеотиддифосфаты (НДФ):
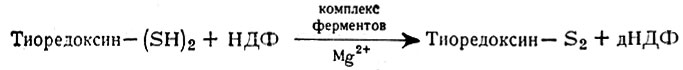
Вновь образованная ДНК и служившая шаблоном ДНК могут на своих концах соединиться под влиянием фермента ДНК-лигазы и образовать циклическую структуру ДНК.
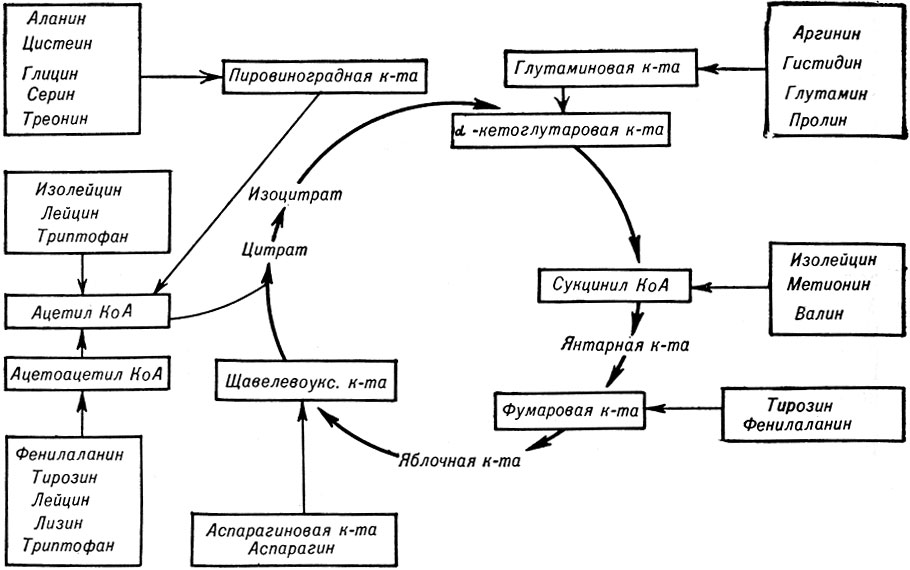
Рис. 6. Цикл трикарбоновых кислот (по Ленинжеру)
Синтез РНК осуществляется при участии полинуклеотидфосфорилазы — фермента, обусловливающего обратимую реакцию соединения нуклеозидднфосфатов в присутствии ионов магния и первоначальной РНК:
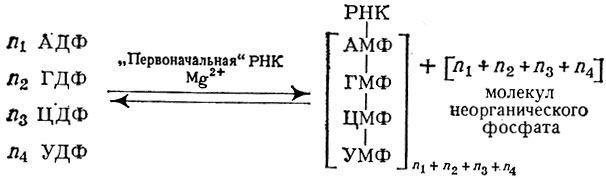
Схема биосинтеза РНК
Образованный полимер содержит 3′-5 ′-фосфодиэфирные связи, к-рые расщепляются рибонуклеазой. Реакция обратима и может быть направлена справа налево (в сторону распада полимера) при увеличении концентрации неорганического фосфата. Первоначальная РНК в данном случае не играет роли шаблона, по к-рому синтезируется полинуклеотид. Скорее всего свободная ОН-группа, находящаяся в концевом нуклеотиде РНК, необходима для присоединения к ней последующих нуклеотидов независимо от входящих в их состав оснований.
По-видимому, в интактной клетке полинуклеотидфосфорилазе принадлежит функция не образования полимера, а расщепления РНК. Что касается высокополимерной РНК с определенной последовательностью нуклеотидов, то образование ее осуществляется РНК-полимеразой, действие к-рой аналогично ферменту, синтезирующему ДНК. РНК-полимераза активна в присутствии ДНК-шаблона, осуществляет синтез РНК из нуклеозидтрифосфатов и собирает их в последовательности, предопределенной структурой ДНК:
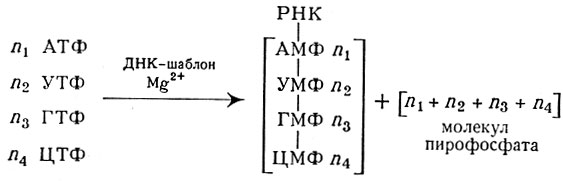
Схема синтеза полимерной РНК
Катаболизм ДНК и РНК
Деградация ДНК и РНК происходит по этапам. Ферменты, расщепляющие РНК, — рибонуклеазы — широко представлены в различных животных тканях. Под влиянием рибонуклеаз двоякого типа — трансфераз и истинных гпдролаз — из РНК образуются олиго- и мононуклеотиды.
Ферменты, расщепляющие ДНК, принадлежат к нуклеазам — гидролазам; результатом их действия является образование олигонуклеотидов как с концевым 5′-фосфатом, так и 5′-фосфатом. Под влиянием диэстераз, специфических нуклеотидаз, фосфорилаз, фосфатаз и нуклеозидаз происходит деградация нуклеотидов с образованием свободных пуриновых и пиримндиновых оснований, дальнейшее превращение к-рых идет по разным путям.
Пуриновые основания — аденин и гуанин — подвергаются гидролитическому дезаминированию под влиянием ферментов аденазы и гуаназы. Из аденина образуется 6-оксипурин (гипоксантин), из гуанина — 2,6-диоксипурин (ксантин). Эти превращения ампнопуринов могут происходить и без предварительного распада соответствующих нуклеотидов и нуклеозидов. Гипоксантин и ксантин подвергаются дальнейшему окислению под влиянием фермента ксантиноксидазы. Конечным продуктом этого окисления является 2, 6, 8-триоксипурин, или мочевая кислота (см.). У человека мочевая к-та дальнейшим превращениям не подвергается и является постоянной составной частью мочи п конечным продуктом обмена пуриновых нуклеотидов и пу-риновых оснований. У большинства млекопитающих мочевая к-та не представляет собой конечного продукта обмена веществ и переходит в аллантоин под действием фермента уриказы.
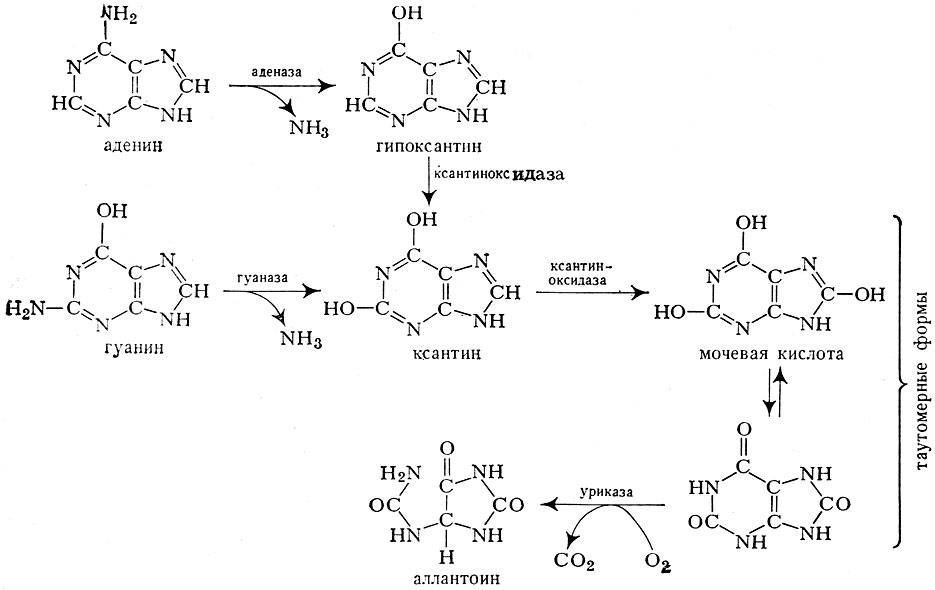
Рис. 7. Схема распада (деградации) пуриновых оснований
Стадии дезаминирования и окисления пуриновых оснований представлены на рис. 7.
По иному пути идет деградация пиримидиновых оснований (см.). Первый шаг заключается в восстановлении урацила в дигидроурацил с последующим гидролизом, приводящим к образованию сначала β-уреидопропионовой к-ты, а затем β-аланина, NH3 и СО2 (рис.8):
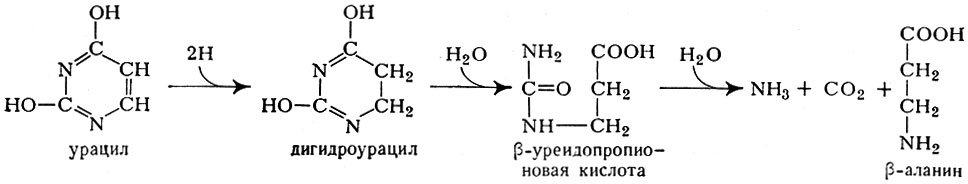
Рис. 8. Схема распада (деградации) пиримидиновых оснований
Аналогичные превращения тимина приводят к образованию β-аминоизо-масляной к-ты.
Т. о., конечным продуктом обмена пуринов у человека является мочевая к-та, а пиримидинов — углекислый газ и аммиак, к-рые могут быть источниками образования мочевины. Что касается (β-аланина, то эта аминокислота участвует в биосинтезе дипептидов карнозина (см.) и ансерина (см.), в большом количестве содержащихся в скелетной мускулатуре позвоночных животных.
В мышечной ткани человека содержится лишь карнозин.
β-аланин является также составной частью пантотеновой к-ты, а следовательно, и коэнзима А, играющего очень важную роль в обмене жирных кислот, стеринов, а также в цикле трикарбоновых кислот.
Регуляция процессов азотистого обмена
А. о. так же, как и все виды обмена веществ, регулируется нервной системой как непосредственно, так и через воздействие на железы внутренней секреции. Основное значение нервной регуляции А. о. заключается в приспособлении его к изменяющимся условиям внешней и внутренней среды. В силу этого выпадение нервных воздействий на органы и ткани приводит к тяжелым нарушениям их структуры и функции.
Благодаря весьма сложным и тонким механизмам регуляции А. о. у взрослого здорового человека обеспечивается относительное постоянство состава азотистых компонентов органов, тканей и внутренней среды организма. Избыток вводимых с пищей азотистых соединений выводится с мочой и калом, недостаток — пополняется из состава тканей тела (это не относится к числу незаменимых соединений).
Насколько состав крови и тканей обладает относительным динамическим постоянством, настолько изменчив состав мочи, отражающий особенности обмена веществ в значительно большей степени, чем состав плазмы крови или цельной крови. В силу этого для выводов об особенностях А. о. необходимо прежде всего знать качественный и количественный состав принимаемой пищи, исследовать качественный и количественный состав азотистых соединений, выделяемых с мочой и калом, и сопоставить полученные данные. Определение особенностей состава крови может дать представление о качественном своеобразии нек-рых сторон А. о., но не позволяет дать заключение о его состоянии в целом. Напр., увеличение содержания белков в пищевом рационе приведет к незначительному повышению содержания небелковых азотистых составных частей (остаточного азота) крови, но выделение азотистых соединений и в первую очередь мочевины с мочой будет значительно повышено. Нарушение окислительного фосфорилирования несколько изменит соотношение содержания креатина и креатинина Р сыворотке крови, однако значительно резче изменится содержание этих соединений и их соотношение в моче (см. Креатин, Креатинурия).
Несмотря на всю сложность и разнообразие реакций, протекающих в организме, выделяемые конечные продукты обмена веществ остаются для данного вида при данном режиме питания качественно более или менее постоянными. Они претерпевают значительные отклонения от нормы при различных патологических состояниях организма.
Радиоизотопное исследование азотистого обмена
Методические приемы изучения отдельных стадий превращения азотистых соединений значительно расширились и обогатились. Большую роль сыграло внедрение в практику исследовании А. о. органических веществ, содержащих радиоактивные или тяжелые изотопы различных элементов и в первую очередь фосфора, углерода, серы, азота, кислорода, водорода. Использование этих соединений позволило довольно детально следить за их превращениями, за постепенным переходом метки из одного вещества к другому, завершающимся выделением изотопа в составе конечных продуктов обмена веществ. В наст. время можно получить практически любую меченую аминокислоту, принимающую участие в процессах биосинтеза белков, свойственных данному организму. Эти эксперименты позволяют выяснить, где и когда аминокислоты включаются в состав белков, с какими веществами или структурами связываются аминокислоты до того, как войдут в состав пептидной цепи. Если аминокислоту глицин, меченную изотопом N15, ввести с пищей в организм животного, то значительная часть изотопа будет быстро выведена из организма в составе мочевины, другая часть остается в тканях и выводится очень медленно. Большая часть введенного препарата с изотопом N15 обнаруживается в белках, причем треть меченого азота включается в белок в виде остатков глицина, а остальные две трети — в составе других аминокислотных остатков. С помощью меченых соединений были открыты или уточнены многие стадии метаболических процессов, идущих в клетках. Так, напр., было подтверждено, что аминогруппы переходят от одной аминокислоты к другой (процесс переаминирования).
Патология азотистого обмена
Патология А. о. проявляется в форме патологии синтеза белков и нарушений обмена различных азотсодержащих метаболитов (аминокислоты, мочевина, аммиак, креатин и креатинин, мочевая к-та и др.), циркулирующих в крови и выделяемых почками.
Основная форма патологии синтеза белков — белковая недостаточность — наступает при нарушении соотношения между процессами биосинтеза и катаболизма белковых структур, приводящем к преобладанию процессов распада над синтезом. Общая белковая недостаточность, характеризующаяся ограничением синтеза многих белков (тканевых., плазменных, ферментных), развивается при алиментарном их дефиците — при общем недоедании и при дефиците энергетических компонентов пищи — углеводов и жиров. В последнем случае белки расходуются в организме в качестве источника энергии (см. Обмен веществ и энергии). Такой же механизм развития общей белковой недостаточности имеет место при нарушении усвоения отдельных пищевых продуктов в связи с патологией пищеварительного аппарата. Ускоренная эвакуация пищи из желудка, а также гипо- и анацидные состояния ограничивают гидролиз пищевых белков, что затрудняет их дальнейшее переваривание. Наиболее выражено нарушение переваривания белков после обширной резекции желудка. Недостаточное расщепление пищевых белков наблюдается также при выпадении действия ферментов сока поджелудочной железы вследствие закупорки или сдавления ее выводящего протока. При энтеритах и энтероколитах усвоение пищевых белков ограничивается из-за ослабления секреторной и ускорения моторной функции тонкой кишки, а также нарушения ее всасывательной способности. При недостаточном питании или питании преимущественно растительными белками и при нарушении усвоения пищевых белков ограничение синтеза разнообразных белков в организме наступает не только из-за количественного недостатка аминокислот, но и из-за нарушения соотношения в содержании отдельных незаменимых аминокислот (дисбаланс). При выраженной белковой недостаточности наступает состояние отрицательного азотистого баланса, при к-ром количество выделяющегося из организма азота больше, чем количество азота, поступающего в организм.
Причиной расстройства белкового обмена в форме усиленного распада является нарушение регуляции метаболизма белковых структур. Ослабление и выпадение нервных воздействий на ткани приводит к нарушению их трофики и развитию трофических язв. Недостаток гормонов анаболического действия (соматотропный гормон, инсулин, половые гормоны) сопровождается первичным ослаблением биосинтеза белков. Недостаток соматотропного гормона у детей вызывает выраженное торможение роста. Дефицит инсулина при некомпенсированном сахарном диабете приводит к преобладанию процессов распада и отрицательному азотистому балансу. Первичное усиление распада белков наблюдается при тиреотоксикозе, избыточном действии стероидных гормонов коры надпочечников.
Усиление распада белков в тканях происходит также при повреждениях тканей (травмы, воспаление, аллергическая альтерация, ишемия, дегенерация). При общей интоксикации, в частности инфекционного происхождения, и обширных травмах мягких тканей и трубчатых костей преобладание распада в метаболизме белковых структур имеет генерализованный характер. Известную роль в этом играют продукты распада, поступающие из поврежденных тканей в общую циркуляцию.
Патология белкового обмена, кроме нарушения соответствия процессов синтеза и распада, в отношении отдельных видов белков проявляется также в форме врожденной недостаточности их биосинтеза, вследствие чего развивается, напр., агаммаглобулинемия (см.), анальбуминемия (см.). Патология обмена белков может быть также в форме извращенного синтеза отдельных видов белков, она проявляется в образовании аномальных по своей структуре белков — нек-рые виды гемоглобинопатии, появление белка Бене-Джонса (см. Бене-Джонса белок), парапротеинов при миеломной болезни (см.) и др.
Патология обмена аминокислот. Патология трансаминирования в форме недостаточности этого процесса возникает при уменьшении активности ферментов — трансаминаз (см. Ферменты), осуществляющих перенос аминогруппы с аминокислоты на α-кетокислоту. Такое нарушение имеет место при алиментарном гипо- или авитаминозе B6, поскольку витамин B6 является предшественником фосфопиридоксаля, а последний представляет собой активную группу (кофермент) трансаминаз. Абсолютной алиментарной недостаточности витамина В6 практически не встречается. Относительная недостаточность его поступления в организм может развиваться при повышенной потребности в нем, напр, при беременности или при значительном подавлении антибиотиками и сульфамидными препаратами нормальной микрофлоры кишечника, где синтезируется витамин В6 в количестве, лишь частично покрывающем суточную потребность организма.
Недостаточность фосфопиридоксаля в организме может развиваться также вследствие нарушения ферментных систем, превращающих витамин B6 в его активную форму (метаболический авитаминоз), что может наблюдаться, по-видимому, при лечении фтивазидом больных туберкулезом.
Уменьшение активности трансаминаз может возникнуть и вследствие нарушения синтеза белковых структур трансаминаз (при белковой недостаточности) или изменения их конфигурации (связывание функциональных групп циклосерином, используемым при лечении туберкулеза). Локальное нарушение трансаминирования в отдельных органах возникает при повреждении их клеточных структур, особенно при некрозе последних. Это сопровождается выходом в кровь внутриклеточных ферментов и повышением в крови активности отдельных трансаминаз, определение к-рых используется в клинике в диагностических целях. Нарушение трансаминпрования в самих поврежденных органах носит при этом комплексный характер, оно обусловлено не только потерей ферментов из клеток, но и нарушением их биосинтеза, в т. ч. и ферментов синтеза фосфопиридоксаля.
Изменение интенсивности процесса трансаминирования в организме происходит и в результате нарушения соотношения реагирующих субстратов. При недостатке α-кетокислот, что может иметь место при угнетении цикла Кребса (напр., при гипоксии, диабете), трансаминирование угнетается, а при избытке аминокислот, наблюдающемся при усиленном распаде белков, трансаминирование может быть усиленным. В последнем случае может возникать вторичное угнетение окисления в цикле Кребса (см. Окисление биологическое, Трикарбоновых кислот цикл).
Фактором нарушения трансаминирования может явиться расстройство регуляции активности отдельных трансаминаз под влиянием гормонов щитовидной железы и коры надпочечников.
Угнетение процесса дезаминирования может наступать в связи с причинами, вызывающими ослабление процесса трансаминирования, т. к. многие аминокислоты быстрее теряют свою аминогруппу в реакции трансаминирования с α-кетоглутаровой к-той, чем в реакции прямого окислительного дезаминирования. Глутаминовая к-та, образующаяся при аминировании α-кетоглутаровой к-ты, быстрее, чем все остальные аминокислоты, подвергается окислительному дезаминированию с образованием аммиака. Этому способствует наличие в клетках специфического фермента — глутаматдегидрогеназы, функционирующей с участием НАД. В ряде исследований показано, что при экспериментальном авитаминозе В6 или при инактивации фосфопиридоксаля тубазидом содержание отдельных аминокислот, кроме глутаминовой, в крови увеличивается, а образование мочевины в печени уменьшается.
Угнетение окислительного дезаминирования в печени возникает также вследствие ослабления биосинтеза белковых структур соответствующих ферментов при белковой недостаточности.
Ослабление окислительного дезаминирования наблюдается также при различных формах гипоксии (геморрагический шок и др.).
Следствием нарушения дезаминирования являются гипераминоацидемия (см. Аминоацидемия) — увеличение доли азота аминокислот в составе остаточного азота (см. Азот остаточный) и даже общая гиперазотемия и аминоацидурия (см.).
Наиболее отчетливо эти сдвиги в А. о. наблюдаются при обширных поражениях клеток печени, особенно при гипоксии органа, когда нарушается не только процесс дезаминирования аминокислот, но и процесс мочевинообразования. При этом в составе остаточного азота, содержание к-рого может значительно возрастать, увеличивается концентрация азота аминокислот, а относительное (или даже абсолютное) количество азота мочевины уменьшается (продукционная гиперазотемия).
Продукционная гиперазотемия возникает и при патологических состояниях, сопровождающихся массивным распадом белков в организме. В этих условиях дезаминирование аминокислот и мочевинообразование в печени могут быть относительно недостаточными, и содержание остаточного азота крови будет увеличиваться за счет свободных аминокислот.
Увеличение содержания остаточного азота происходит также и при нарушении выделительной функции почек. Однако в этих условиях гиперазотемия происходит гл. обр. за счет увеличения в крови концентрации мочевины (ретенционная гиперазотемия). Клинической формой выраженной ретенционной гиперазотемии является уремия (см.). Гиперазотемии могут иметь и смешанный генез при одновременной недостаточности функции почек и печени и усиленном распаде белков. Продукционная гиперазотемия, не осложненная нарушением выделительной функции почек, приводит к потере аминокислот организма с мочой, т. к. фильтрация аминокислот в клубочковом аппарате почек превышает в этих условиях возможности их реабсорбции в канальцах (см. Почки). Усиленное выведение аминокислот выявлено в условиях белкового голодания при раневом истощении, травматических повреждениях трубчатых костей, спинного и головного мозга, в тяжелых случаях ожога, при инфекционных заболеваниях, в стадии кахексии при злокачественных новообразованиях, при гипертиреозе, болезни Иценко—Кушинга, длительном лечении глюкокортикоидами и препаратами АКТГ. В этих случаях гипераминоацидурия отражает в основном относительную недостаточность процессов дезаминирования избыточно освобождающихся при распаде белка аминокислот. Не исключено, что при этих состояниях может иметь место и прямое угнетение процессов дезаминирования в отдельных тканях, особенно в печени.
Другая группа гипераминоацидурий объединяет различные по происхождению формы нарушения обмена аминокислот, при к-рых увеличение их выделения связано с нарушением реабсорбции в канальцевой системе почек.
Генерализованное нарушение реабсорбции аминокислот происходит при фильтрации их из крови не в свободном состоянии, а в комплексе с металлами. Показано, что аминокислоты крови легко образуют комплексы с медью, свинцом, кадмием, ураном и при этом выводятся из организма.
При болезни Вильсона — Коновалова или гепато-лентикулярной дегенерации (см. Гепато-церебральная дистрофия), для к-рой характерно нарушение обмена меди, наблюдается значительная экскреция аминокислот в комплексе с медью без одновременного увеличения концентрации аминоазота в крови.
Нарушение почечной реабсорбции аминокислот имеет место и при синдроме Фанкони (см. Цистиноз), называемом некоторыми авторами аминовым диабетом. Для этого заболевания характерно сочетание усиленного выведения аминокислот (количество аминоазота в моче увеличивается в 30—40 раз) с гиперфосфатурией и псевдорахитическими изменениями в костях. Наблюдается также почечная глюкозурия (см. Диабет почечный).
Избирательное нарушение реабсорбции известно в отношении цистина. Однако цистинурия (см.) обычно сопровождается общим нарушением обмена этой аминокислоты. Описана врожденная аномалия обмена цистина, проявляющаяся в резко выраженной цистинурии без повышения содержания цистина в крови. Выведение цистина с мочой достигает в этих случаях 400—800 мг в сутки, тогда как нормальная экскреция цистина не превышает 80 мг. Цистин сравнительно плохо растворим, и увеличение его экскреции сопровождается образованием цистиновых камней в мочевыводящих путях.
Более тяжелое нарушение обмена цистина известно под названием цистиноза (см.). Это заболевание сопровождается общей аминоацидурией, в том числе и цистинурией, отложением кристаллов цистина в элементах ретикулоэндотелиальной системы; при нем наблюдается ранний смертельный исход.
Угнетение превращения фенилаланина в тирозин относится к наследственным заболеваниям. В крови и моче значительно увеличивается количество фенилаланина и ряда промежуточных продуктов его обмена, в частности фенилпировиноградной и фенилуксусной кислот. Клинически это нарушение обмена проявляется значительным отставанием умственного развития — фенилпировиноградная олигофрения (см. Фенилкетонурия). Недостаточно полное превращение фенилпировиноградной и фенилуксусной кислот в фенилацетилглутамин — нормальный конечный продукт обмена той части фенилаланина, к-рая не подвергалась превращению в тирозин, — выявлено и при вирусном гепатите. Ограниченное образование фенилацетилглутамина в этих случаях обусловлено первичным ограничением образования глутамина в печени.
Нарушение окислительного превращения тирозина в конечные продукты его обмена (фумаровую и ацетоуксусную кислоты) может сопровождаться накоплением различных промежуточных продуктов. Так, нарушение первого этапа этого пути обмена (переаминирования с α-кетоглутаровой к-той) приводит к гипертирозинемии, тярозинурии и состоянию тирозиноза (см.). Этот механизм нарушения обмена выявлен при экспериментальной белковой недостаточности, поражении печени четыреххлористым углеродом и экспериментальном лейкозе у мышей. В клинике аналогичное нарушение обмена тирозина наблюдается у больных лейкозом и при коллагенозах. Другая форма нарушения обмена тирозина — алкаптонурия (см.), развивающаяся при задержке окислительного превращения тирозина на стадии гомогентизиновой кислоты (см.). Патология относится к врожденным аномалиям обмена.
Нарушение других направлений в обмене тирозина также связано с активацией или угнетением ферментов, катализирующих реакции его специфических превращений. Превращение тирозина через стадию ДОФА в пигменты (меланины), окрашивающие кожу и волосы, определяется активностью тирозиназы, представляющей собой специфический медьсодержащий белок. Активность тирозиназы регулируется меланофорным гормоном гипофиза, синтез к-рого сдерживается гормонами коры надпочечников. При гипофункции надпочечников могут возникать нарушения пигментного обмена (см.). Альбинизм (см.) представляет собой врожденную аномалию обмена тирозина, состоящую в выпадении синтеза фермента тирозиназы.
Основной путь обмена триптофана в организме заканчивается превращением его в никотиновую к-ту. Ряд промежуточных продуктов на этом пути обмена триптофана, а именно 3-оксикинуренин, ксантуреновая, 3-оксиантраниловая кислоты и их производные, обладают при повышенной концентрации патогенными свойствами.
Ксантуреновая к-та способствует распаду гликогена и гипергликемии. При длительном повышении ее концентрации в крови у экспериментальных животных наблюдаются дегенеративные изменения в бетаклетках поджелудочной железы. 3-Оксикинуренин и 3-оксиантраниловая кислоты могут проявлять также канцерогенное действие.
Накопление в крови промежуточных продуктов обмена триптофана происходит вследствие подавления активности ряда ферментов, функционирующих в комплексе с производными витаминов В6, B1, В2 и PP. Избыточное образование токсических метаболитов выявлено при хроническом гепатите, тяжелых формах сахарного диабета, остром лейкозе, хронических миело- и лимфолейкозе, лимфогранулематозе, ревматизме и склеродермии. Нарушение обмена триптофана может быть выявлено при помощи пробы с нагрузкой.
Увеличение концентрации креатинина в крови происходит при нарушении выделения его почками, а увеличенное пли уменьшенное выведение его с мочой, без одновременной ретенции в крови, отражает нарушение образования его из креатина при патологии обмена последнего в мышечной ткани. Увеличение экскреции креатинина наблюдается при гипофункции щитовидной железы. Уменьшение экскреции креатинина в сочетании с увеличенным выведением креатина имеет место при гипертиреозе, тяжелом течении сахарного диабета и особенно при миопатиях (миастения, миозит, миотония).
Количество азота мочевой к-ты — конечного продукта пуринового обмена (см.) — в составе остаточного азота крови колеблется от 0,1 до 3.0 мг%. Патологическое увеличение его концентрации наблюдается при массивном распаде клеточных структур (голодание, тяжелая мышечная работа, инфекции и т. д.), когда усиление эритропоэза (см. Кроветворение) сопровождается освобождением ядер из ретикулоцитов. Выведение мочевой к-ты с мочой ограничивается ее интенсивной реабсорбцией. Увеличение концентрации мочевой к-ты в крови обусловливает возможность отложения ее в хрящах, суставных сумках, сухожилиях, фасциях, а иногда в почках, мышцах и коже.
См. также Наследственные болезни.
Азотистый обмен в облученном организме
Характер изменений А. о. в основном зависит от дозы облучения. При воздействии больших доз ионизирующей радиации в организме происходит процесс патологического распада белков органов и тканей, к-рые не восстанавливаются белками пищи, что проявляется в отрицательном азотистом балансе, особенно при облучении в летальных дозах.
В изменении А. о. в облученном организме существенную роль играет пониженное всасывание аминокислот стенками тонкой кишки, а также повышенное выделение азота с мочой в ближайшие дни после лучевого поражения. Так, напр., при тотальном воздействии гамма-излучением и нейтронами выделение аминокислот с мочой у людей увеличивается в 10 раз по сравнению с нормой. При облучении в больших дозах экспериментальных животных отмечалось увеличение содержания мочевины в моче, тирозина в крови, выведение аминокислот с мочой, креатинурия (см.), что указывает на усиление тканевого распада. Увеличение распада белков может быть также результатом повышения активности протеолитических ферментов. В свою очередь повышение активности протеолитических ферментов связывают с непосредственным поражением внутриклеточных мембран.
Изменения А. о. при облучении зависят от следующих основных причин: непосредственное воздействие радиации на молекулы белка в клетке и изменение его физ.-хим. свойств; изменение биохимических механизмов синтеза белка; интенсификация протеолитических ферментов в клетке; опосредованное влияние радиации на деятельность желез внутренней секреции и т. д.
Изменение азотистого обмена в процессе старения
В пожилом возрасте существенно снижается функциональная способность пищеварительного тракта (ослабляется синтез и секреция соляной к-ты, протеолитических ферментов), замедляется всасывание свободных аминокислот в кишечнике; снижается способность ассимиляции пищевых веществ на тканевом и клеточном уровнях, что связано в первую очередь с дизадаптацией ферментных систем организма; нарушаются процессы биосинтеза белков, нуклеиновых кислот и т. д.
По мере старения организма снижается его способность ассимилировать белки, увеличиваются эндогенные потери белковых компонентов пищи, что характеризуется появлением отрицательного азотистого баланса.
Причины снижения интенсивности синтеза белка в старости до сих пор остаются неясными. Большинство исследователей считает, что при старении первичные изменения возникают в регуляторных генах, приводя в одних случаях к нарастающему подавлению транскрипции отдельных оперонов (см.), а в других — к временному усилению биосинтеза нек-рых белков. При этом неравномерно изменяется биосинтез различных белков, сокращается возможный диапазон активации биосинтеза, быстрее нарастает снижение потенциальных возможностей биосинтетических систем в условиях напряженной деятельности. В последующем наступают изменения и в структурных генах, что приводит к определенным качественным сдвигам в синтезируемых белковых молекулах, в частности к изменениям в аллостерическом регулировании активности ферментов.
Изменение содержания и активности ферментов, состояния белков мембран клетки и субклеточных структур ведет к существенному нарушению процессов образования, накопления и использования энергии в клетке, что в свою очередь приводит к снижению уровня биосинтетических процессов.
Характерным примером изменения А. о. при старении является нарушение пуринового обмена, когда в крови и тканях накапливается большое количество уратов, к-рые отлагаются затем в суставах и хрящах (см. Подагра). Однако так наз. отложения солей связаны не только с нарушениями пуринового и минерального обмена.
Имеются убедительные доказательства, что причины отложения солей в суставах и хрящах заключаются не только в увеличении концентрации уратов и кальция, а прежде всего в изменении свойств структурных белков соединительной ткани, в частности коллагена. Качественные изменения белков проявляются гл. обр. в нарушении третичной и четвертичной структуры молекулы белка (см.). При этом отмечено повышение прочности белковой структуры, обусловленное появлением дополнительных, перекрестных связей между отдельными компонентами. В процессе старения меняются физ.-хим. свойства белков, в частности снижается лабильность, дисперсность, гидрофильность и электрический заряд их молекул. Гипотеза отечественных авторов (А. А. Богомолец, А. В. Нагорный, В. Н. Никитин) о важности для процессов старения изменения физ.-хим. свойств белков, напр, белков соединительной ткани, их огрубения и понижения функциональной активности, находит все большее признание в мировой литературе.
Многочисленные экспериментальные и клинические наблюдения особенностей разбалансирования процессов А. о. при старении послужили основанием для разработки специальных рационов. В основе рекомендаций для составления таких рационов, направленных на нормализацию нарушений А. о. у лиц пожилого и старческого возраста, лежат: принцип энергетической сбалансированности пищевого рациона с энерготратами организма; обеспечение в рационах сравнительно высоких количеств белка (1,2—1,3 г на 1 кг веса) с высоким содержанием полноценных животных белков (гл. обр. белков молока); ограничение в рационах продуктов высокой концентрации пуриновых оснований (см.); обеспечение в рационах достаточного содержания витаминов и микроэлементов, в частности аскорбиновой к-ты, ниацина, тиамина, рибофлавина, кобаламина и др., что необходимо для обновления изнашивающихся в процессе жизнедеятельности ферментных систем.
Особенности азотистого обмена у детей
Интенсивность процессов А. о. на протяжении роста ребенка подвергается изменениям, особенно ярко выраженным у новорожденных и детей раннего возраста. В течение первых трех дней жизни баланс азота является отрицательным, что объясняется недостаточным поступлением белка с малым количеством пищи. В этот период обнаруживается транзиторное повышение остаточного азота в крови до 55—60 мг% . Количество выделяемого почками азота нарастает в течение первых 3 дней, после чего падает и начинает вновь увеличиваться со второй недели жизни параллельно возрастающему количеству пищи.
Общая особенность А. о. у детей — положительный баланс азота, что является необходимым условием роста. Азот пищи в максимальной степени используется растущим организмом для пластических целей. Так, напр., на ранних этапах развития детского организма ферментные системы, обеспечивающие синтез нуклеиновых кислот, отличаются наивысшей активностью, в то же время активность ферментов, катализирующих их распад, снижена.
Наиболее высокая усвояемость азота в организме наблюдается у детей первых месяцев жизни. Баланс азота заметно снижается в период 3—6 мес. жизни, хотя п остается положительным.
Во втором полугодии жизни баланс азота стабилизируется. По данным В. Ф. Ведрашко (1958), у детей 2—3 лет, получающих 4—4,2 г/кг белка, баланс азота составляет 2,3 г, ретенция (т. е. задержка) — 30% при соотношении животных и растительных белков 4:1. У детей 4—6 лет удовлетворительный баланс и ретенция азота достигаются при получении 3,5 г/кг белка: баланс 2,7 г, ретенция — 25% (В. Ф. Ведрашко и Э. И. Аршавская, 1965). У детей 7—8 лет азотистое равновесие достигается при введении 2,5 г/кг белка: баланс 2,8—3 г, ретенция — в пределах 21% . По данным Ин-та питания АМН СССР, у детей 11—13 лет при введении 2 г/кг белка азотистый баланс составляет 1,8 г, ретенция — 13,8%.
Показатели ретенции и баланса азота подвержены значительным индивидуальным колебаниям, зависят от количества белка пищи, его соотношения с другими пищевыми ингредиентами. Установлены также сезонные колебания этих показателей: они выше в весеннее и летнее время и ниже зимой.
Потребность в незаменимых аминокислотах у детей выше, чем у взрослых, при этом для детского организма к незаменимым аминокислотам относят и гистидин. Средние величины потребности в незаменимых аминокислотах, по данным ФАО ВОЗ(1963), представлены в таблице 3.
| Аминокислота | Грудные дети (мг/кг веса в день) | Дети школьного возраста (мг/кг веса в день) |
|---|---|---|
| Гистидин | 34 | - |
| Изолейцин | 119 | 30 |
| Лейцин | 150 | 45 |
| Лизин | 103 | 60 |
| Метионин | 45 ( |
27 ( |
| Фенилаланин | 90 ( |
27 ( |
| Треонин | 87 | 35 |
| Триптофан | 22 | 7,4 |
| Валин | 105 | 53 |
Клетки растущих тканей отличаются высокой концентрацией аминокислот, что свидетельствует о высокой активности механизмов, обеспечивающих транспорт аминокислот через клеточные мембраны. Активный мембранный транспорт аминокислот имеет место в плаценте. Дент (С. Е. Dent, 1948) говорит в связи с этим о «плацентарном аминокислотном насосе», обеспечивающем движение аминокислот от матери к плоду (см. Плацента). Показано, что этот процесс отличается строгой стереоспецифичностью, т. е. левовращающие (L-аминокислоты) проходят плацентарный барьер с более высокой скоростью, чем правовращающие (D-аминокислоты). Функция плаценты позволяет объяснить более высокое содержание аминокислот в пуповинной крови по сравнению с кровью детей более старшего возраста и взрослых (табл. 4).
| Аминокислота | В пуповинной крови | В крови детей | В крови взрослых |
|---|---|---|---|
| Алании | 4,8 | 3,9 | 3,8 |
| Аргинин | 3,3 | 2,2 | 2,1 |
| Глицин | 3,4 | 2,6 | 2,8 |
| Гистидин | 3,4 | 1,8 | 1,7 |
| Изолейцин | 2,3 | 1,7 | 1,6 |
| Лейцин | 2,5 | 2,3 | 2,0 |
| Лизин | 8,1 | 2,4 | 2,8 |
| Метионин | 0,5 | 0,3 | 0,35 |
| Фенилаланин | 2,3 | 1,6 | 1,6 |
| Треонин | 2,8 | 2,3 | 2,0 |
| Триптофан | 1,7 | 0,8 | 1,1 |
| Тирозин | 2,3 | 1,6 | 1,4 |
| Валин | 4,9 | 3,2 | 3,0 |
Существенное влияние на рост ребенка оказывают дефекты питания, вследствие к-рых ребенок получает избытки отдельных аминокислот, что служит причиной задержки физического развития, гипераминоацидурии, интоксикации.
У детей раннего возраста повышена экскреция аминокислот с мочой — так наз. физиологическая гипераминоацидурия. На первой неделе жизни азот аминокислот составляет 3—4% от общего азота мочи (по нек-рым данным, до 10%) и лишь к концу первого года жизни снижается до 1 %. В этот период выведение аминокислот в расчете на 1 кг веса достигает величин выведения их у взрослого человека, экскреция аминоазота, достигающая у новорожденных 10 мг/кг, на втором году жизни редко превышает 2 мг/кг. В моче новорожденных повышено по сравнению со взрослыми содержание таурина, треонина, серина, глицина, аланина, цистина, лейцина, тирозина, фенилаланина и лизина. В первые месяцы жизни в моче обнаруживается также этаноламин и гомоцитруллин. В моче детей первого года жизни преобладают аминокислоты пролин и гидроксипролин. Причиной физиологической гипераминоацидурии является функциональная незрелость почек, проявляющаяся в недостаточной реабсорбции аминокислот из клубочкового фильтрата (гипераминоацидурия ренального типа). Доказательством этого служит более высокий клиренс аминокислот. У недоношенных, кроме того, имеет место гипераминоацидурия перегрузочного типа, т. к. содержание свободных аминокислот в плазме крови выше, чем у доношенных.
В процессе роста ребенка меняются количественные и качественные характеристики А. о.
Еще Вирхов (R. Virchow, 1856) обратил внимание на то, что моча плода содержит избытки мочевой к-ты и лишь следы мочевины. Отложение кристаллов мочевой к-ты в почечной ткани он назвал мочекислым инфарктом новорожденных. Исследования важнейших азотистых компонентов мочи у детей показали, что соотношение мочевой к-ты, мочевины н аммиака в процессе роста существенно изменяется. Так, первые 3 месяца жизни характеризуются наименьшим содержанием в моче мочевины и наибольшей экскрецией мочевой к-ты. В возрасте от 3 до 6 мес. в моче нарастает количество мочевины и снижается содержание мочевой к-ты. Выведение мочевой к-ты на протяжении первого — второго года жизни в расчете на 1 кг веса превышает таковое у взрослых, содержание аммиака в моче в первые дни жизни невелико, но затем резко возрастает и держится на высоком уровне на протяжении всего первого года жизни. Н. Ф. Толкачевская (1960) связывает эти особенности А. о. с преобладанием у плода и новорожденного урикотелического пути обмена аммиака (нейтрализация аммиака обеспечивается гл. обр. за счет усиленного образования мочевой к-ты). Это филогенетически более древний путь, к-рый на первом году жизни постепенно и почти полностью вытесняется уреотелическим — синтезом мочевины в цикле Кребса—Гензелейта.
Важной особенностью А. о. у детей является физиологическая креатинурия (см.). Креатин обнаруживается в амниотической жидкости и в моче, начиная с периода новорожденности и до периода полового созревания. Суточная экскреция креатинина с возрастом увеличивается, в то же время по мере нарастания массы тела азот креатинина в процентном отношении к общему азоту мочи снижается. Количество выведенного с мочой креатинина в расчете на 1 кг веса у детей колеблется в пределах 5,5—10 мг. Величины суточного выведения креатинина, процентного отношения к общему азоту мочи, полученные у разных детей одного возраста, близки между собой.
Библиогр.: Белозерский А. Н. Молекулярная биология — новая ступень познания природы, М., 1970; Браунштейн А. Е. Биохимия аминокислотного обмена, М., 1949, библиогр.; Збарский Б. И., Иванов И. И. и Мардашев С. Р. Биологическая химия, Л., 1972; Иванов И. И. и др. Введение в клиническую биохимию, Л., 1969, библиогр.; Химия и биохимия нуклеиновых кислот, под ред. И. Б. Збарского и С. С. Дебова, Л., 1968; Lehninger A. L. Biochemistry, N. Y., 1970.
Радиоизотопное исследование А. о. — Белки, под ред. Г. Нейрата и К. Бэйли, пер. с англ., т. 3, ч. 2, с. 594, М., 1950, библиогр.; Хаггис Д ж. и др. Введение в молекулярную биологию, пер. с англ., с. 341, М., 1967.
Патология А. о. — Ангелов А. М. и др. Влияние тироксина на активность некоторых ферментов обмена углеводов и аминокислот в печени морских свинок, Вопр. мед. хим., т. 17, в. 2, с. 165, 1971, библиогр.; Бадалян Л. О., Таболин В. А. и Вельтищев Ю. Е. Наследственные болезни у детей, с. 44, М., 1971; Капланский С. Я. Вопросы патологии обмена белков и аминокислот, в кн.: Хим. основы процессов жизнедеятельности, под ред. В. Н. Ореховича, с. 253, М., 1962, библиогр.; он же, Патологическая физиология белкового обмена, Многотомн. руководство по пат. физиол., под ред. Н. Н. Сиротинина, т. 2, с. 455, М., 1966, библиогр.; Кремер Ю. Н. Биохимия белкового питания, Рига, 1965, библиогр.; Лаптева Н. Н. Патофизиология белкового обмена, М., 1970, библиогр.; Майстер А. Биохимия аминокислот, пер. с англ., с. 463, М., 1961; Мардашев С. Р. и д р. Опыт и перспективы лечения L-глютамином больных тяжелым эпидемическим гепатитом (болезнь Боткина), в кн.: Усп. гепатол., под ред. Е. М. Тареева и А. Ф. Блюгера, с. 401, Рига, 1971; Репин И. С. Азотистый обмен при лихорадочных состояниях, Л., 1961, библиогр.; Xорст А. Молекулярная патология, пер. с польск., с. 196, М., 1967, библиогр.
А. о. в облученном организме — Бак З. и Александер П. Основы радиобиологии, пер. с англ., М., 1963, библиогр.; Ранние радиационно-биохимические реакции, под ред. Е. Ф. Романцева, М., 1966, библиогр.; Штреффер К. Радиационная биохимия, пер. с нем., М., 1972.
Изменение А. о. в процессе старения — Ведущие проблемы советской геронтологии, под ред. Д. Ф. Чеботарева и др., Киев, 1972; Ведущие проблемы возрастной физиологии и биохимии, под ред. В. Н. Никитина, М., 1966, библиогр.; Парина Е. В. Возраст и обмен белков, Харьков, 1967, библиогр.; Фролькис В. В. Регулирование, приспособление и старение, Л., 1970, библиогр.; Gsеll D. Protein and nitrogen metabolism in the old age, Proc. 4-th int. congr. dietetics, p. 88, Stockholm, 1965; Verz&##225;r F. Aging of the collagen fiber, Int. Rev. Connect. Tissue Res., v. 2, p. 243, 1964.
А. о. у детей — Питание здорового И больного ребенка, под ред. М. И. Олевского и Ю. К. Полтевой, с. 59, 212, М., 1965; Толкачевская Н. Ф. Развитие процессов обмена у детей первого года жизни, М., 1960, библиогр.; Тур А. Ф. Физиология и патология новорожденных детей, Л., 1967; Schreier K. Eiweiβstoffwechsel, Handb. d. Kinderheilk., hrsg. v. H. Opitz u. P. Schmid, S. 57, B. u. a., 1965, Bibliogr.; Sсhreiеr К. u. a. Über die Clearance-Rate einiger Aminosäuren bei Säuglingen und Frühgeborenen, Z. Kinderheilk., Bd 79, S. 165, 1957, Bibliogr.; SereniF. a. Principi N. The development of enzyme systems, Pediat. Clin. N. Amer., v. 12, p. 515, 1965, bibliogr.
Источники:
- Большая медицинская энциклопедия. Том 1/Главный редактор академик Б. В. Петровский; издательство «Советская энциклопедия»; Москва, 1974.- 576 с.
|
ПОИСК:
|
При копировании материалов проекта обязательно ставить активную ссылку на страницу источник:
http://sohmet.ru/ 'Sohmet.ru: Библиотека по медицине'